
 首页
首页 登录
登录 注册
注册




 下载:
下载:
-
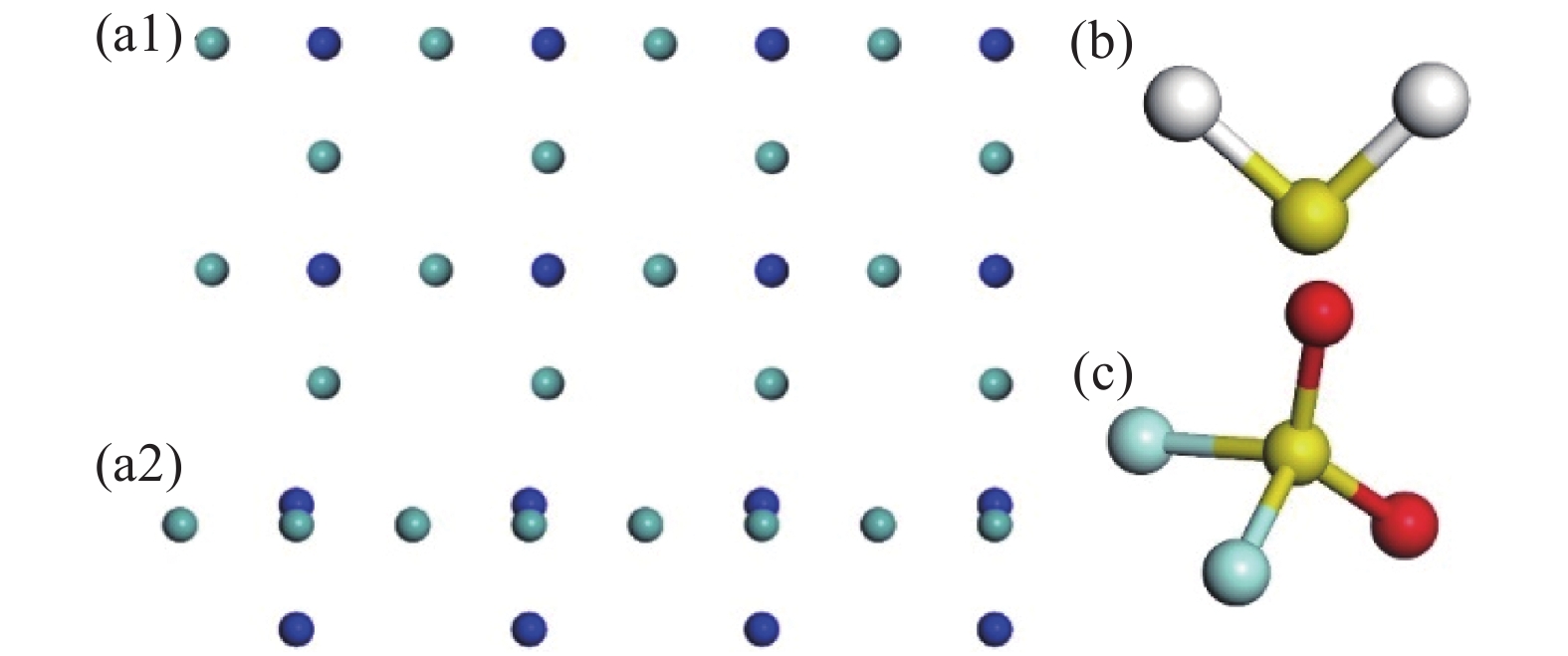
纳米传感器因其体积小,检测精度高,成本低的优点,成为未来绝缘电气设备内置传感,在线监测的发展方向。作为纳米传感器的关键部分,纳米材料发展至今,有较多性能优异的典型代表。例如过渡金属硫化物、半导体性质的金属氧化物,过渡金属氮化物、碳化物等[1]纳米材料,因其材料特质在纳米传感器领域受到广泛关注。与这些广为人知的二维结构的纳米材料不同,作为一种过渡金属氮化物纳米材料,因为基于Mo3N2的薄膜所具有的优越的物理、化学和机械性能已被研究用于各种应用之中[2]。因Mo3N2具有优异的催化性能,气体分子较易吸附在其表面并改变其导电性能,且Mo3N2材料对外几乎呈现金属性,即能耗较低的特质,为其作为器件设计及生产提供可能。
SF6,一种无毒不燃的稳定气体,因具有优异电负性,已成熟应用于各气体绝缘设备之中[3],但这些电力设备在运行过程中出现的局部放电或者局部过热会导致绝缘介质被击穿,发生分解,产生系列可恢复的低氟化物,但由于设备长期运行后,密封性降低而增加的微水、微氧等成分与这些低氟化物发生反应,最终产物为几种成分较为稳定的化合物[4-5]。这些气体的存在将进一步促进SF6的分解,最终降低电力设备中绝缘介质含量,影响设备工作性能。在GIS中,H2S,主要分解产物之一,含量较多,而SO2F2作为一种典型剧毒分解产物,不仅对设备运维人员的构成安全隐患,还会腐蚀电力设备内壁[6-7]。及经长期的设备运行监测及相应实验研究发现,通过对这些气体组分的检测,可以实现对SF6绝缘状态及电力设备运行状态的评估[8-9]。因此,采取一些检测手段对这些分解产物的含量进行检测是有必要的[10]。在以下几个部分中,本文将对上述两种分子在Mo3N2上的吸附性能进行理论计算,从吸附前后对吸附能、电荷转移、能带结构、态密度(分波态密度)这几个方面进行对比,分析Mo3N2的传感机理。对Mo3N2作为气敏材料的可行性研究提供数据支持。
全文HTML
-
本文使用Materials Studio(MS)商业软件中的Dmol3软件包完成全部的理论计算。在本文的仿真计算中,对交换关联泛函的处理选用了广义梯度近似(GGA)以解决电子密度不均的现象造成的带隙计算不准问题。同时,Perdew-Burke-Ernzerhof(PBE)函数被选用为处理交换关联能[11]。使用Tkatchenko和Scheffler开发的色散校正的密度泛函理论(DFT-D)方法以及TS方法用于理解弱相互作用和范德华力[12]。双数值极化基组(DNP)作为原子轨道基组[13]。对于本文中4×4×1超胞进行几何优化及电子计算过程中,静态电子结构计算中布里渊区MonkhorstPack网格k点设置为2×2×1。几何优化计算中,能量容差精度设为1.0×10−5 Ha,最大力和最大位移分别为0.002 Ha/Å和0.005 Å[14]。为消除吸附模型层间相互作用的影响,真空层设置成15 Å,在所有仿真计算中选用5.0 Å 的全局轨道截止半径和0.005 Ha的拖尾值用以提高收敛速度,并确保总能量的准确性。
Mo3N2对单个气体分子的吸附能定义为:
其中
$E_{({\rm{Mo}}_3{\rm{N}}_2 + {\rm{gas}}) }$ 表示气体分子吸附在Mo3N2晶体上的总能量,$ E_{{\rm{Mo}}_3{\rm{N}}_2} $ 表示未吸附气体分子时原本晶体结构的能量,Egas表示单个气体分子的能量。如果Eads为负值,则说明该吸附过程是自发进行的[15]。表一给出了Mo3N2的最稳定吸附位上的吸附能、吸附距离及电荷转移情况,电荷转移用Q表示,Q大于0表明电荷从吸附基底转移到气体分子上,如果小于0则情况相反[16]。
-
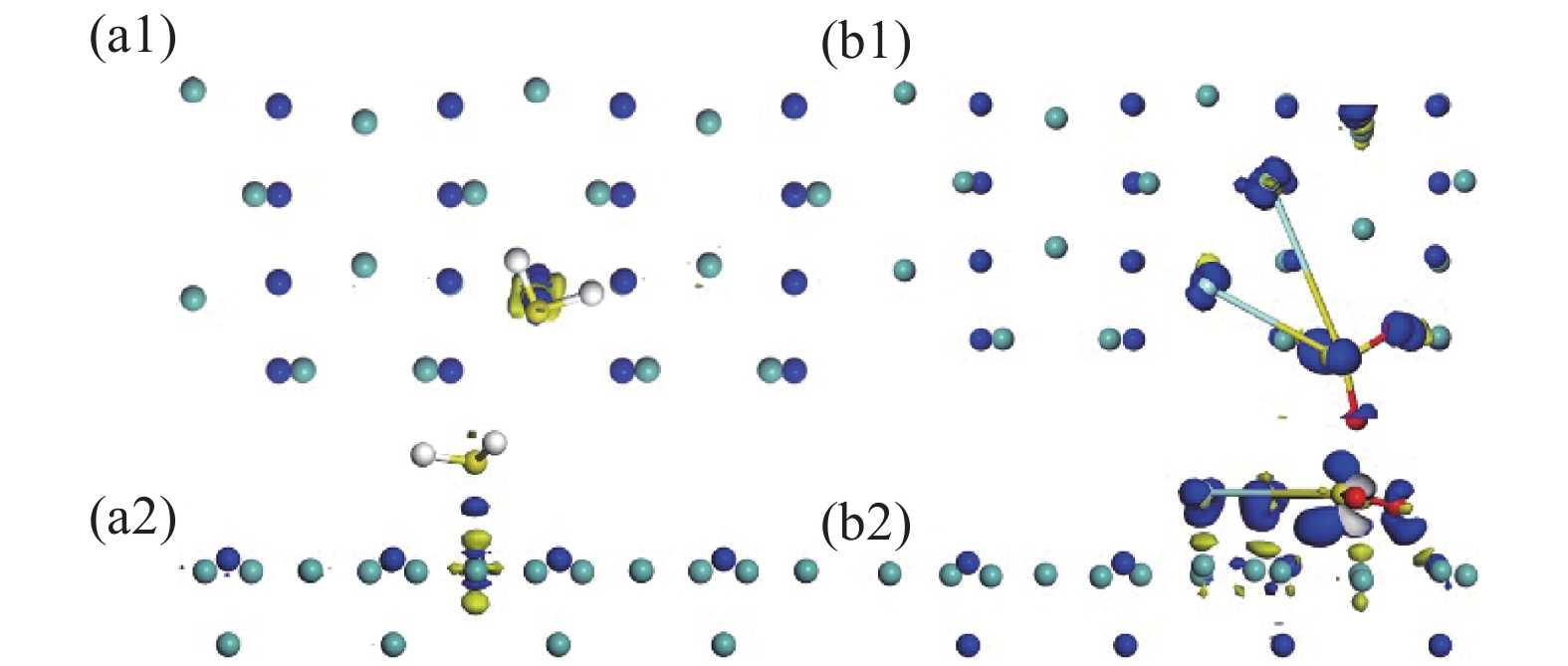
Mo3N2(200)为两层吸附原子排列而成,如图1(a)所示,其中(a1)为Mo3N2俯视图,(a2)为侧视图。从图中可知N原子被Mo原子环绕其中,呈四方型排布。根据这种特殊原子分布情况,本文参照石墨烯的吸附位点,设置了N原子顶部,Mo原子顶部,及Mo-Mo桥位这三种吸附位点,并充分考虑H2S,SO2F2分子的对称性,设置了多种角度及朝向的吸附构型。两种气体的最稳定吸附构型的吸附能、吸附距离、转移电荷及能带带隙情况如表1所示。其中吸附距离是指气体分子距离吸附基底最近的距离。
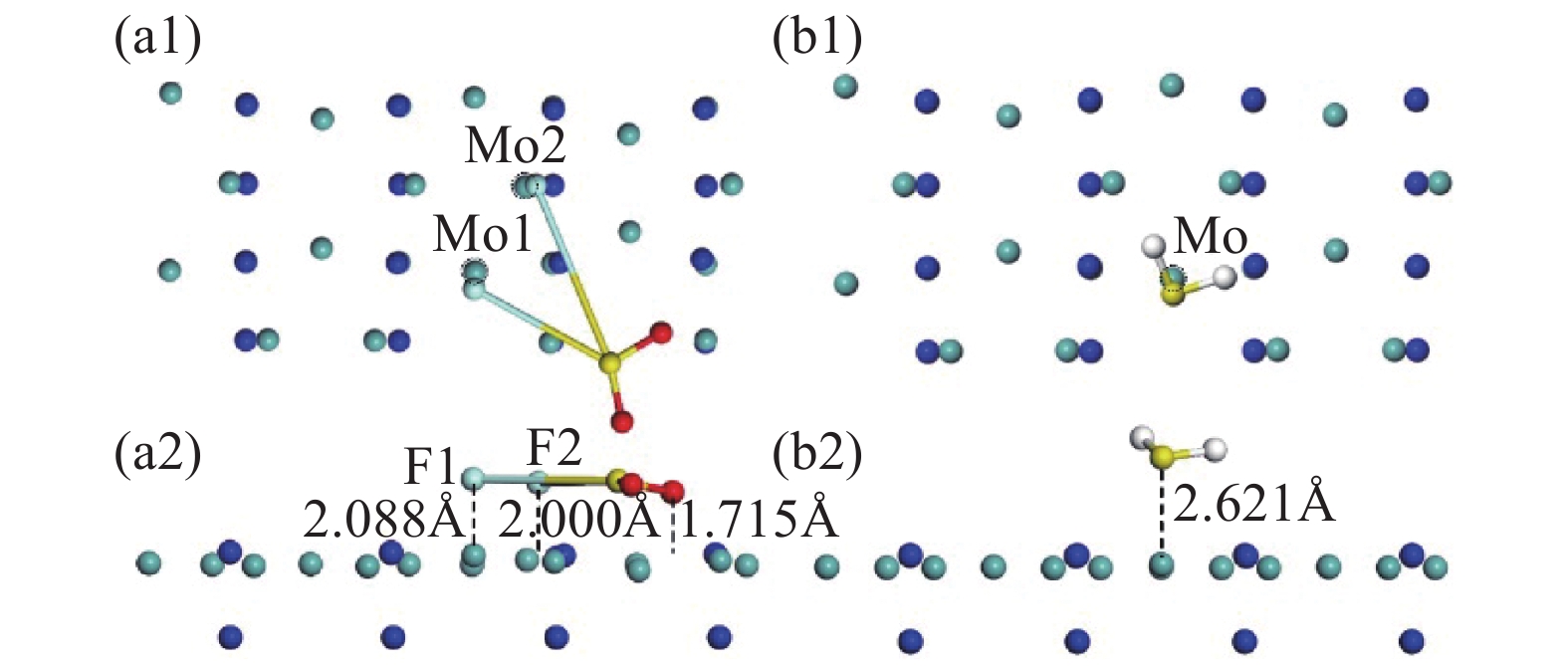
图2展示了Mo3N2对两种气体分子的最佳吸附构型,图2(a)、(b)分别为SO2F2及H2S的最佳吸附构型,从图中可以看到,SO2F2气体分子由原始的一个O原子朝上,两个F原子及另一个O原子呈三角朝下的吸附形态变成5个原子呈平面形态吸附在Mo3N2表面。其中Mo3N2表面的Mo原子发生偏移,有别于原始形态,且F1-S键长为4.120 Å,F1-S键长为5.123 Å,明显长于原始的1.614 Å,可判定S-F键断裂,且两个F原子均向Mo原子移动,考虑该体系吸附能为−6.244 eV,可综合判定该吸附为强烈的化学吸附。H2S吸附体系与SO2F2体系明显不同,虽然Mo3N2表面的Mo原子也发生偏移,但H2S分子无明显形变,吸附距离为2.621 Å,H2S由原始的H原子朝下呈一字型的吸附形态变为整体稍平行于Mo3N2表面,吸附能为−1.958 eV,有0.148 e电荷从Mo3N2表面转移到H2S气体分子,有别于SO2F2吸附体系。
-
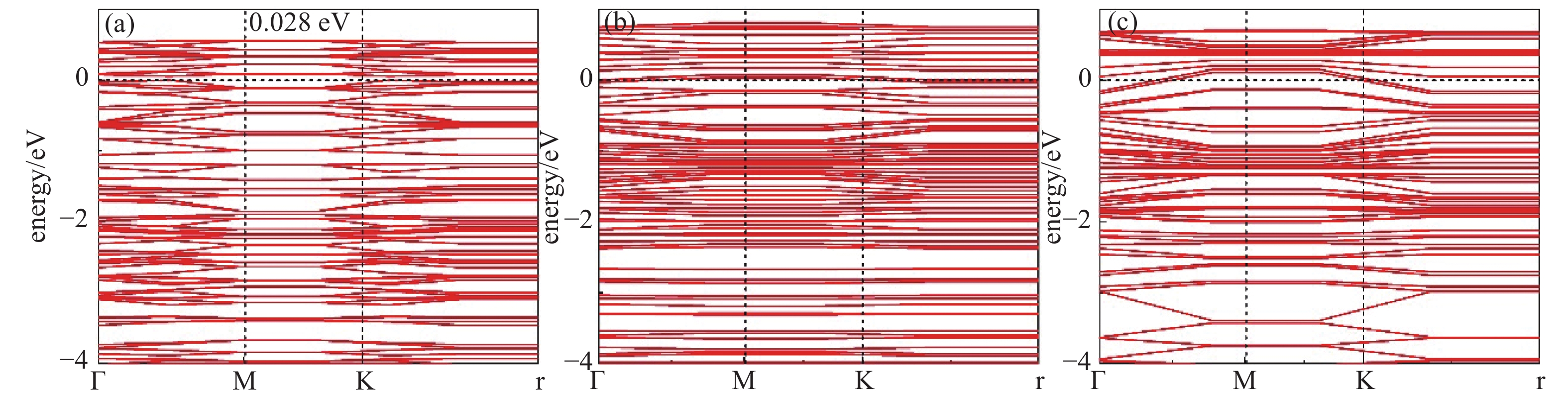
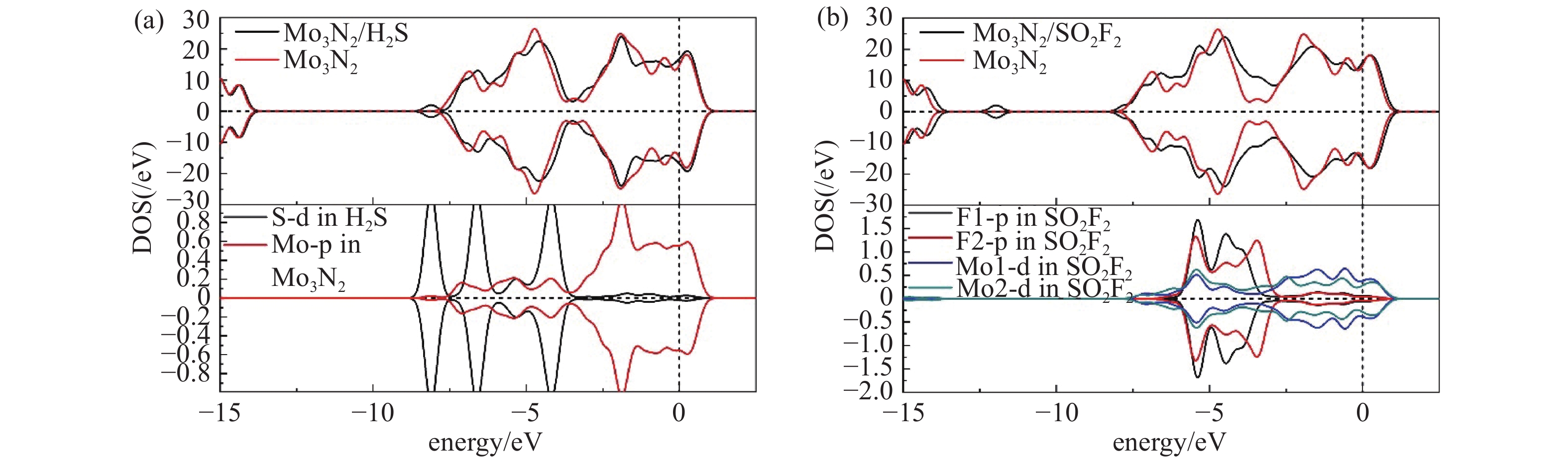
为了更细致的了解这两种气体分子在Mo3N2表面上的吸附行为,进一步绘制了能带图与自旋电子能态密度图(Density of States, DOS),从它们的变化来对比分析吸附气体的变化机理与原因。从图3可以看出Mo3N2表面与两种气体分子吸附完成后的能带结构图变化。图3(a)为Mo3N2的带隙结构,带隙为0.028 eV,即带隙及其微小,呈半导体性质。吸附两种气体后,带隙均变为0,呈金属性[17]。从图4中可以看到两种气体分子在Mo3N2表面吸附前后的自旋态密度图,均呈严格对称分布,即均为无磁性体系,在本节的后续部分不再赘述。图4(a)中可以明显看出,在费米能级±1.000 eV范围内吸附前后的态密度几近完全重合,在−1.000 eV之后吸附体系较Mo3N2体系态密度变化更为平缓,幅值有略微增长,峰值均出现在−2.500、−4.850 eV左右,无明显偏移,吸附后体系较吸附前DOS有所降低。可以看出这个−2.500 eV处,未被占据的DOS峰主要是由离气体分子最近的Mo原子的P轨道贡献所得,而−4.850 eV处的未被占据的DOS峰则由S原子的d轨道与吸附基底中离H2S分子最近的Mo原子的P轨道贡献所得(后续所讨论的Mo原子均为离气体分子最近的吸附基底中的原子)。在费米能级附近可以看出这两个原子轨道无轨道重叠,即相互作用微弱。
从图4(b)中可以看出SO2F2在Mo3N2表面上吸附前后的能态密度在导带处基本重合,而在价带处出现不同程度的偏移。吸附气体后,导带0.500 eV、价带1.000 eV出现的未被占据的峰主要是由Mo1原子和Mo2原子的p轨道杂化贡献所得。在价带3.500 eV处,两个F原子的p轨道分别与其最近的Mo原子的d轨道交叠,相互作用明显。在费米能级出,两种原子交叠程度明显高于SO2,且分子间原子重叠,轨道杂化明显,结合SO2F2分子裂解,两个F原子远离分子主体的吸附行为,印证其吸附性质为吸附能较大的化学吸附[18]。
图5展示了两种气体分子在Mo3N2表面上完成吸附后最稳定模型的差分电荷密度,非常形象的展现了分子与晶体间的电荷转移情况。通过CDD计算公式
$\Delta \rho \text{=}\rho ({\rm{Mo}}_3{\rm{N}}_2+{\rm{gas}})-\rho {\rm{Mo}}_3{\rm{N}}_2\text{-}\rho {{\rm{gas}}}$ 可以计算出每种气体分子最稳定吸附模型的CDD,其中$\Delta \rho \text{=}\rho ({\rm{Mo}}_3{\rm{N}}_2+{\rm{gas}})$ 为整个吸附模型的电荷密度,$ \rho {\rm{Mo}}_3{\rm{N}}_2 $ 则为Mo3N2的电荷密度,$\rho {{{\rm{gas}}}}$ 为一个气体分子的电荷密度,需要注意的是,以上几个电荷密度必须为同一模型内的计算数据[19]。H2S吸附在Mo3N2表面上CDD如图5(a)所示,可以清楚看出Mo原子附近聚集了主要的电荷积累,些许电荷积累同样存在与吸附基底与气体分子之间,电荷减少出现在吸附基底的Mo原子周围,可知Mo与S原子之间存在较强的电荷局域化行为。以Hirshfeld法得知,H2S在吸附后转移了0.148 e电子到Mo3N2表面,表明H2S分子在反应中的失电子性能。SO2F2吸附在Mo3N2表面上CDD如图5(b)所示,可以清楚看出电荷主要积累在SO2F2周围,电荷减少则出现在吸附基底与SO2F2分子周围,可知SO2F2与吸附基底之间存在较强的电荷局域化行为。SO2F2在吸附后转移了从Mo3N2表面转移了
0.7927 e电子,表明SO2F2分子在反应中的得电子性能。上述电荷转移描述再次验证了对于两种SF6分解组分气体而言,Mo3N2晶体表面对SO2F2气体的吸附能力优于H2S。 -
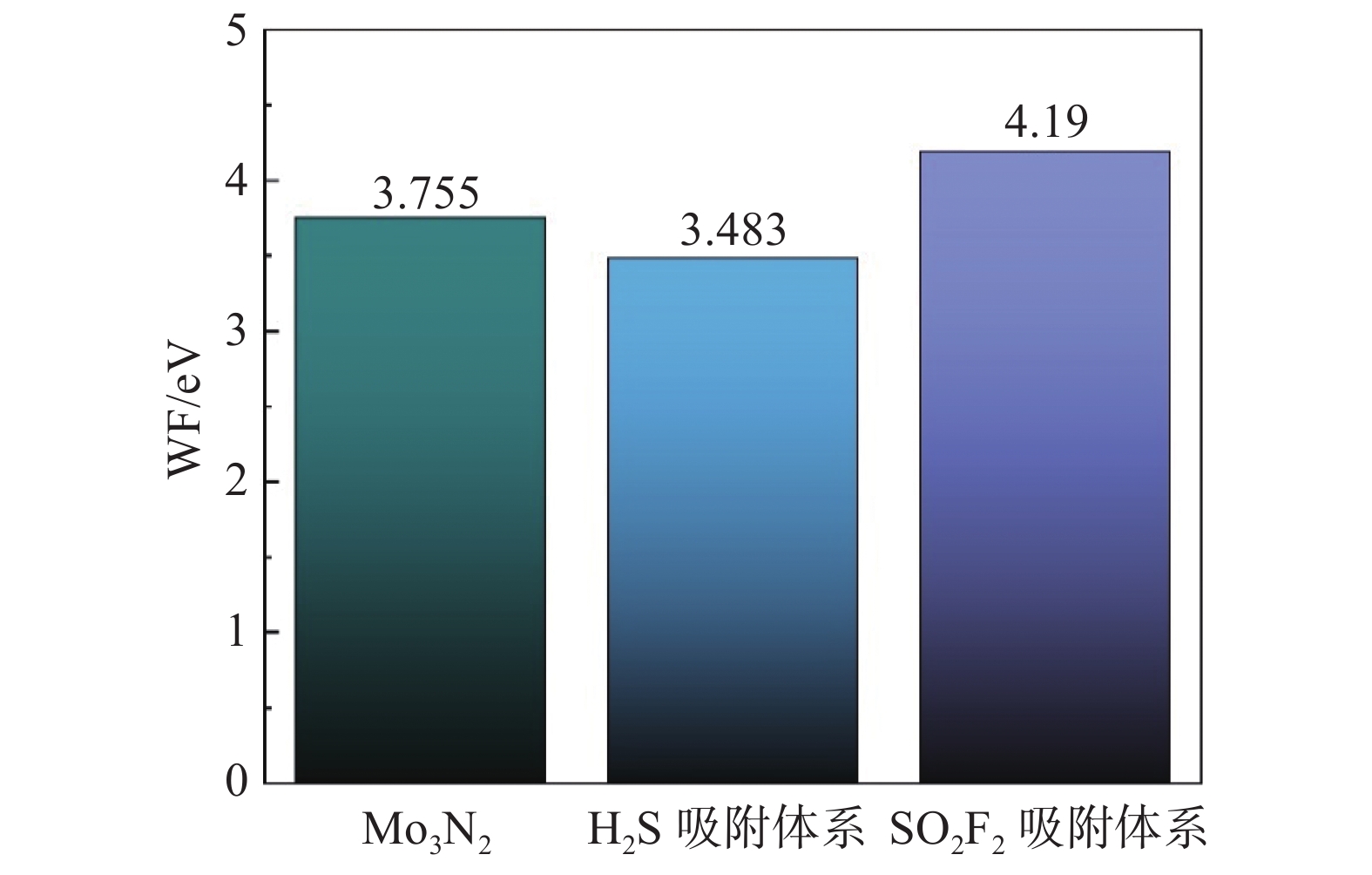
功函数(Work Function,WF)表达了一个电子从费米能级跨跃到真空层中所需的最少能量。换种说法而言,功函数可以被看做在气体在吸附过程中触及吸附基底,发生电荷转移的壁垒。本节为Mo3N2在场效应传感器领域的扩展应用提供理论数据支撑[20]。
图6展示了Mo3N2以及H2S和SO2F2吸附体系的功函数大小。从图中可以看出,Mo3N2体系的功函数为3.755 eV,在吸附了单个H2S分子以后,Mo3N2的功函数降低为3.483 eV,而吸附SO2F2后,整个体系功函数增加至4.190 eV。这表明吸附单个SO2F2分子后,电子从吸附基底转移的过程变得不易,吸附H2S后与电子转移难易程度则与之相反。这两个相反的功函数数值变化进一步证实了在Mo3N2场效应管传感器领域,可以通过调节栅极电压的形式来控制传感器电阻变化规律[21],以达到对气体的检测目的。
2.1. 最稳定吸附构型
2.2. 电子性质
2.3. 功函数分析
-
本文通过对SF6的两种主要分解组分的基于第一性原理的MS仿真计算,得出了两种气体分子在Mo3N2表面上的最稳定吸附模型,及其相应的吸附能,吸附距离、转移电荷等数值。从结果可以得出以下结论:
(1)两种气体的吸附改变Mo3N2的带隙,使其从半导体状态转变为金属性;
(2) Mo3N2对H2S,SO2F2的吸附能分别为−1.958 eV、−6.244 eV,结合Hirshfeld法进一步分析电荷转移情况确定对单个H2S,SO2F2的吸附行为均为化学吸附;
(3)通过对能带结构及能态密度分析得出Mo3N2有潜力作为电阻型气敏传感器对两种气体进行气敏检测,而WF分析表明其在场效应传感器对气体进行选择性检测方面的可行性;
(4)SO2F2在吸附在Mo3N2表面的过程中因强相互作用导致S-F键断裂,表明Mo3N2具有作为SO2F2清除剂的潜力。
本文所有研究结果为开发Mo3N2基气敏传感器在电力设备故障检测领域的应用上提供理论支持。