
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
膜结构为生命活动划定了边界, 而膜上的通道蛋白通过精确调控营养物质与代谢物质、离子及信号分子的跨膜转运, 帮助细胞维持内环境稳态, 更为生命活动塑造了各种复杂的功能. 如电压门控钠离子通道能够感受膜电位并改变自身构象, 介导钠离子流帮助心肌细胞产生动作电位[1]; 也有机械力敏感钙离子通道mPiezo1, 在细胞膜张力影响下改变结构从而介导钙离子信号进入细胞, 帮助细胞识别机械力信号[2]; 还有氯离子转运蛋白CLC家族, 其CBS结构域影响着其转运活性, 进而调节溶酶体等细胞器的功能活性[3]. 对通道蛋白结构-功能的研究会帮助研究者深入理解这些复杂的生理功能, 为攻克重大疾病提供许多潜在药物靶点, 也为合成生物学构建能够控制物质输入与输出的人工细胞提供极为重要的参考. 传统的研究方法往往只能从群体水平揭示通道蛋白的平均行为, 单分子技术的出现彻底打破了这一局限, 凭借其高灵敏度、高时空分辨率的独特优势, 从分子定位、轨迹追踪、构象标定、结构动力学研究、功能测定等不同维度为人们打开了微观世界的大门[4–6]. 其中, 荧光技术和膜片钳技术作为两类重要的单分子研究手段, 在生物膜上各类分子的运动和结构动力学以及功能发挥的研究中展现了独特价值和关键见解[5,7]. 膜片钳技术[8,9]是电生理学领域极具开创性意义的实验技术, 通过对微小区域内通道蛋白电流的精确测量, 测定通道蛋白的功能. 凭借这一技术, 科研人员得以深入探究神经元的兴奋性、心肌细胞的电活动以及各类细胞离子通道相关疾病的发病机制[7,10]. 膜片钳技术中, 细胞贴附式记录是一种十分重要的细胞原位记录模式, 凭借其高分辨率特性, 能够精准捕获细胞原位上单个通道在不同生理条件下的开放概率、离子导通率等关键特性[11–15], 为理解通道蛋白的功能奠定了基础. 荧光技术在生物研究中应用极为广泛, 借助荧光标记, 科研人员可以利用荧光显微镜对单个通道或通道群体进行可视化观察[16–19]. 荧光共振能量转移(FRET)技术利用两个荧光团之间的能量转移现象[16,19–21], 为监测通道蛋白的构象变化提供了有效手段, 近年来更是发展出了一系列工具, 如SIFA技术[22,23]、queenFRET技术[24]、lipoFRET技术等[25], 对膜通道蛋白的结构动力学研究贡献了多维度的技术支持.
不同的单分子技术在单独应用时存在各自的局限性, 而生命活动是一个多维度、多层面相互交织的复杂动态过程, 研究者需要结合不同单分子技术才能够完整剖析一个通道蛋白的详细工作过程[14,26,27]. 细胞中较高的背景荧光限制了膜片钳钳制区域的成像分辨率[28], 单分子成像体系中膜片钳技术往往无法获得良好的封接, 种种因素限制了二者在通道蛋白功能结构解析上的联用. 通过对不同条件下单独测量的荧光信号和电流信号动力学模式进行匹配, 目前结合单分子荧光技术和膜片钳技术已经实现了许多突破性的进展, 例如CFTR有关研究, 依靠相同条件下独立进行的单通道电生理实验与单分子荧光实验的统计特征匹配, 识别了不同构象所具备的不同功能[27], 却未能够实现直接观测结构与功能之间的同时变化. 或有依靠特殊的荧光标记配体有限地实现了原位联合记录[29,30], 但在配体未结合时无法获取蛋白结构信息, 荧光标记配体的模式更难以普适地帮助到其他需要原位联合记录的研究.
为了突破这些局限, 本文提出一种选择性局域激发光路设计, 在细胞膜上表面实现了范围可控的局域照明模式, 并能够在照明区域内进行单分子荧光成像与动态追踪. 依托光路中可调的反射镜与光阑, 照明光斑能够进行平面移动与尺寸调节, 达成激发成像区域与玻璃电极共定位, 进而有效地整合膜片钳技术. 选用中心光阑并调整聚焦位置可以进一步提高选择性, 通过削减无关区域的照明强度, 有效克服细胞内高背景荧光, 实现小范围高信噪比的单分子荧光成像, 以针对性地适配细胞贴附式膜片钳技术. 这些技术提供了一套简便高效的细胞膜表面单分子荧光成像方案, 为蛋白质结构-功能耦联研究开辟了新的技术路径. 荧光膜片钳耦联技术将能够帮助确认单通道记录区域中, 寡聚通道蛋白的化学计量数以及通道蛋白与功能相关的结构动力学信息.
-
实验中使用拉针仪P97(美国Sutter公司生产), 将玻璃毛细管(脑赛思公司生产, 型号为150N86F, 全长10 cm, 内径0.86 mm, 外径1.50 mm, 两头抛光, 含导液丝)制作成针尖. 使用参数为HEAT = 500, PULL = 0, VEL = 50, DEL = 1. 完成针尖拉制后, 使用与固化剂以10∶1重量比混合的聚二甲基硅氧烷(PDMS, SYLGARD 184)对针尖颈部进行涂覆, 操作中注意不能让PDMS堵住针尖口. 在85 ℃下经过2 h的烘烤, PDMS固化附着在针尖上. 最后使用锻针仪(Narishige, MF2)进行锻针, 将针尖靠近铂丝并加热直至针尖微微融化回缩, 使得针尖失去较为锐利的断口, 有利于提高单通道记录的稳定性.
-
选择性局域激发光路的主要控制组件为可控反射镜、光阑与凹透镜(图1(a)): 根据整体光路确定凹透镜焦距, 关键标准为到达物镜后焦面的激光是否平行, 并以此为基准允许其沿光轴方向微调移动, 文中凹透镜的焦距为50 cm; 光阑可选用最典型的孔径光阑与中心光阑, 其中中心光阑的遮挡半径根据扩束后的激光直径设计, 所使用的中心光阑直径为6 mm. 实验采用的滤片依照荧光分子而定, 本文使用的Cy3对应的滤镜组为ZET532/640 x(EX), ZT532/640 rpc(DM), ZET532/640(EM).
在实验中, 通过光路调整可以改变细胞膜上表面单分子荧光成像的范围. 沿光轴方向对凹透镜位置进行调节, 以此改变聚焦光斑距离物镜的高度, 令聚焦光斑略高于焦面, 使其到达没有背景荧光的细胞浴液中. 焦面上会形成一片较大的照明区域(相较于光斑尺寸), 调节孔径光阑的孔径大小, 可以改变焦面上照明区域的大小. 对于高信噪比单分子成像实验而言, 调节凹透镜位置使得聚焦光斑恰好位于焦面并使用中心光阑替换孔径光阑.
-
将100 pmol/L的胆固醇-DNA-Cy3添加在细胞浴液中, 孵育5 min后, 更换细胞浴液为无荧光浴液, 此时细胞膜外表面将被标记上Cy3荧光.
-
将HEK293 (Human Embryonic Kidney Cells, HEK293)细胞放置于MEM培养基(Thermo Fisher公司生产, Gibco系列)中进行传代, 培养基中添加10%胎牛血清(Fetal Bovine Serum, FBS)和1%青霉素、链霉素(P&S), 培养条件为37 ℃, 5% CO2. 使用Lipo3000 (Thermo Fisher公司生产, 货号L3000015)将相应的质粒(即外源基因)转染至细胞中, 使HEK293细胞能够表达目标蛋白. 细胞用于膜片钳实验之前, 需要按照实验设计更换培养基为所需的细胞浴液, 对于细胞贴附式记录模式, 细胞浴液可以统一采用有助于维持细胞状态的Live Cell Image Solution (Thermo Fisher公司生产, 货号A59688DJ).
-
单通道记录前, 使用次氯酸钠溶液处理电极银丝, 形成Ag/AgCl电极. 向针尖中灌注实验所需的电极液, 保证其液面能在针尖安装后接触到电极. 确定细胞位置后, 关闭宽场照明打开激光光源, 通过目镜寻找表达报告荧光蛋白的细胞.
将膜片钳放大器的参考电极放置到细胞浴液中, 保持稳定. 在针尖夹持器上安装准备好的针尖, 施加正压后操纵MP285微操作机械臂将针尖放入细胞浴液, 入液后电阻通常在3—7 MΩ. 操纵针尖轻轻接触细胞, 施加负压形成高阻封接. 对于单通道记录, 通常认为10 GΩ以上是合适的, 符合标准即可开始进行细胞贴附式单通道记录.
完成封接后使用膜片钳放大器商家提供的软件Elements Data Reader3进行记录, 选择200 pA量程, 20 kHz采样率进行信号记录.
对于mPiezo1的电生理测试实验而言, 在表达了mPiezo1的HEK293细胞上形成高阻封接之后, 开启数据记录. 对于Yoda1激活实验, 不做压强调整; 对于压强激活实验, 通过气压组件调整压强后标记施加压强的时间, 以方便后续数据处理.
-
在对齐之前需要确保高阻封接处于EMCCD的成像范围内, 这通常通过封接前控制整个目标细胞处于EMCCD成像范围内来实现. 完成封接后, 通过宽场照明确认针尖所处的位置, 调节光路中反射镜方向对聚焦光斑位置进行调整, 使得光斑对准单通道记录区域.
-
在膜片钳技术中, 根据不同的记录模式, 玻璃电极钳制范围的直径从1 μm到20 μm不等[31,32], 往往只能够接触细胞膜的上表面. 考虑到常见的照明方式与成像模式难以在膜片钳记录区域进行单分子荧光成像, 本工作开发了一种在细胞膜上表面开展单分子荧光成像的方法.
我们对照明光路进行改良, 通过光路中的孔径光阑与凹透镜, 实现细胞膜上表面的低背景局域照明, 进而实现细胞膜上表面, 即膜片钳记录区域的单分子成像. 在具体操作上, 调整凹透镜位置使照明光束经过物镜后聚焦于物镜前焦面的上方, 如图1(a)所示. 此时, 物镜前焦面的一定范围内能受到激光照明, 利用羧基荧光素溶液中荧光分子的匀质性, 我们得以实现对照明光斑光场的成像, 并得到其光场的分布特性(图1(b)).
在实际实验过程中, 借助物镜对焦面进行控制, 能够让膜片钳记录区域被成像光斑照亮. 与此同时, 膜片钳记录区域与成像焦面完美重合, 从而确保成像效果达到最佳状态. 但在膜片钳技术中, 一旦完成高阻封接, 针尖在各个方向的移动会对记录质量造成不利影响. 实验通过微调光路中的反射镜M1(图1(a)), 实现聚焦光斑在物镜前焦面上的平移, 允许在成像光斑与膜片钳记录区域未完全对齐时进行微调. 改变反射镜M1的俯仰角将直接调整聚焦光斑的位置, 如图1(c)右上图所展示的一样, 聚焦光斑将会在焦面上产生直线位移, 图1(c)右下组图中蓝色位移为操作俯仰角所产生的实际效果. 若沿特定轴线(图1(a)红轴线)转动反射镜M1, 光斑直线位移方向会发生变化. 如图1(c)左下图所示, 旋转角ρ的调节将会使得聚焦光斑以圆周进行运动, 图1(c)右下图中红色位移为此操作所产生的实际效果. 总结来说, 结合可调光路, 可以实现聚焦光斑的精准微调. 在焦面上成像光斑的行为可以被分解为两个极坐标参数(r, β), 这两个坐标参数分别可以通过M1的俯仰角θ和旋转角ρ进行调节. 这一方法极大地提升了成像光斑对准膜片钳记录区域的便利性. 图1(d)展示了在细胞贴附式记录中, 针尖带有荧光的情况下, 光斑与针尖从未对齐到对齐, 再到移动过度的过程, 呈现其在实际操作中调节光斑的情况.
-
通过物镜调焦, 让物镜前焦面与细胞膜上表面对齐(图2(a)), 实现上表面的单分子荧光成像与单分子信号追踪, 调整孔径光阑的孔径可以改变照明范围. 同样利用羧基荧光素溶液测定光斑的光场分布情况. 图2(b)展示了大孔径光阑产生的范围照明的光场分布、单分子成像效果以及单分子信号数据. 将其归一化后, 光强超过90%的区域选定为单分子成像区域, 并用该区域的直径来表征成像区域大小. 实验表明, 调节孔径光阑尺寸, 能够改变细胞膜上成像区域的大小, 见于图2(b)—(d)的上半部分.
用100 pmol/L的Cy3荧光标记胆固醇-DNA分子对细胞膜进行单分子级别的染色, 开展细胞膜上表面单分子荧光成像, 由此能获取细胞膜上表面胆固醇-DNA单分子荧光图像, 并在光斑范围内对自由扩散的单分子信号进行追踪, 具体实验结果见于图2(b)—(d)的下半部分. 不同孔径的光阑所提供的照明直径存在差异(图2(e)), 同时还会对采集到的单分子荧光信号质量产生影响(图2(f)). 本文选择采用信号强度与信号标准差的比值作为单分子信号的信噪比(SNR). 越小的成像范围中所记录的单分子荧光信号的信噪比越高. 这种成像方法为细胞膜上表面的单分子荧光成像、追踪提供了可能, 也为类似于巨膜片钳技术、钳制范围较大的膜片钳实验方法提供了钳制区域单分子荧光成像的能力.
-
膜片钳技术中非常重要的细胞原位记录模式——细胞贴附式记录, 其钳制区域通常只有1—2 μm, 对光学成像而言, 由于衍射现象的存在, 荧光分子在钳制区域里的扩散行为是无法被观察到, 且没有意义的. 将细胞原位膜片钳技术与单分子荧光成像技术联用所面临的关键挑战之一, 在于细胞内源性荧光背景的高噪声干扰, 这严重限制了单分子检测的灵敏度与定位精度[28]. 虽然提高激发光强可暂时改善信噪比, 但随之增强的光毒性(phototoxicity)[33,34] 会引起细胞应激反应(如膜电位漂移、蛋白质错误折叠等), 进而干扰生理状态下的分子行为研究. 本工作将采用聚焦照明模式使得光照强度集中于我们所关心的区域, 与此同时, 借鉴片光照明原理, 严格控制光照在细胞中的强度和照明区域, 以此实现成像质量的提升, 有效降低背景干扰, 并最大程度减轻光照毒性对细胞造成的不良影响.
我们进一步改良照明系统, 通过集成凹透镜和中心光阑实现空间选择性的单分子激发, 达成细胞贴附式记录区域的单分子荧光成像. 通过调整凹透镜令到达物镜后焦面的激光为平行光, 再由物镜将平行光汇聚于物镜的前焦面, 形成聚焦光斑, 参照图3(a). 借助这一设计, 光照强度在聚焦点也就是细胞贴附式记录区域达到峰值, 在满足我们关注区域照明需求的同时, 有效降低细胞整体接受的光辐照量. 照明激光 “空心化” 通过在扩束后的激光光路中设置中心光阑实现, “空心”照明光束不会影响聚焦效果, 还能够大幅减少对胞内区域的照明, 显著降低胞内背景荧光的激发. 在成像光路中, 细胞贴附式记录区域附近的离焦荧光, 对成像质量的影响远大于其他区域, 空心照明有效抑制了这部分干扰. 从细胞层面来看, 空心照明光束减小细胞接受辐照的范围, 进一步减轻光毒性, 为细胞提供了更适宜的实验环境, 提升成像质量的同时, 保障细胞生理状态的稳定.
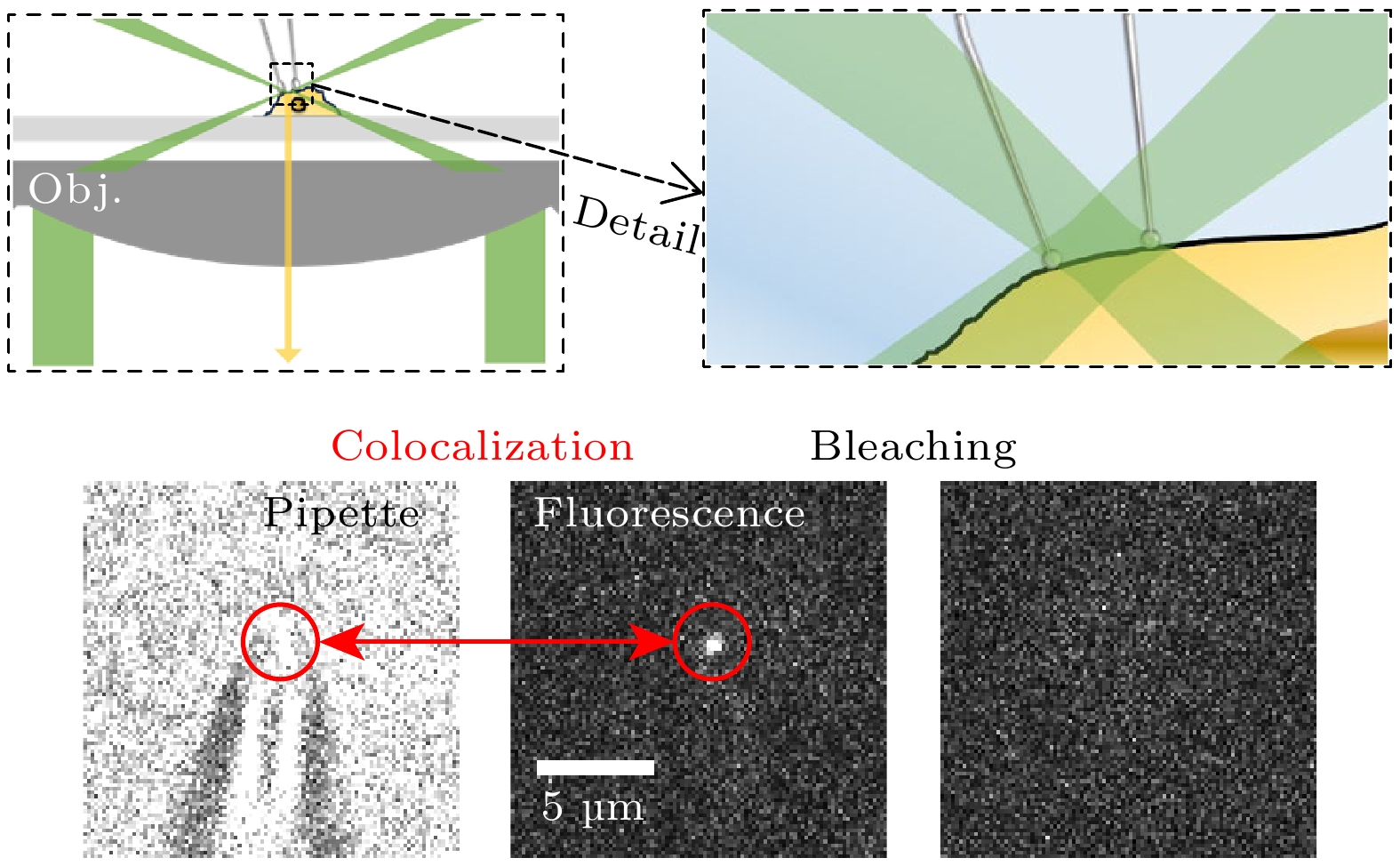
借助羧基荧光素溶液可以确定聚焦光斑在焦面处实现了良好聚焦, 大于峰值照明强度90%的范围直径约为2 μm, 这与细胞贴附式记录所使用的针尖尺寸高度匹配. 得益于聚焦光斑的优质特性, 在该区域能获得足够出色的成像质量. 在细胞膜上表面极小范围内, 能顺利实现高质量的单分子荧光成像(图3(b)), 其信噪比高于所有的范围成像模式. 荧光信号与针尖封接区域的明场成像有着良好的共定位, 且针尖中的荧光信号的漂白过程也能够被显著观察(图3(c)).
-
单通道记录作为一项极为精密的测量技术, 其测量能力极易受到诸多外部环境因素的影响. 在 完成荧光光路改装后, 针对膜片钳系统的信号采 集能力展开了一系列表征测试. 具体实验过程中, 在成功实现高阻封接的区域开展光照测试. 将聚焦光斑对准单通道记录区域, 通过对比光照前后空白电信号的变化情况, 来评估荧光光路改装对高阻封接的影响, 相关数据及结果可参考图4(a). 从电信号数据和统计分析结果来看, 照明操作不会对高阻封接造成影响. 这一特性为单通道记录的稳定性和准确性提供了有力保障, 极大地提升了单通道记录在荧光成像环境下的可行性.
完成光照对高阻封接影响的测试后, 在光照环境下, 借助离子通道 mPiezo1 开展单通道记录测试. 如图4(b)所示, 电极液中配置30 μmol/L Yoda1, Yoda1是一种特异性激活mPiezo1的配体[35], 随后在转染了mPiezo1的HEK293细胞上进行高阻封接, 开始单通道记录. 采集到的典型单通道数据如图4(b)所示, 添加了Yoda1的实验中出现了明显的mPiezo1的电信号, 与文献[35]报道一致, 同时和没有Yoda1的对照组形成了显著对比.
mPiezo1本身是一个机械力敏感的离子通道, 当膜张力增加时, mPiezo1将会被激活[13,35]. 根据图4(c)的实验示意, 在该实验中, 利用针尖中的负压变化来改变钳制区域的膜张力, 进而实现对钳制区域通道的激活. 实验结果表明, 对未转染mPiezo1细胞的记录, 不会出现信号(图4(d)); 而转染了mPiezo1细胞上, 在不同的压强条件下, mPiezo1呈现出不同的开放频率(图4(e)). 具体表现为, 随着压强的逐渐增大, mPiezo1的开放频率也随之增加; 而当施加的压力被释放时, mPiezo1能够迅速关闭.
-
本文围绕照明光路展开改良, 成功达成细胞膜上表面的范围单分子荧光成像与单分子轨迹追踪, 能够与巨膜片钳技术搭配形成荧光膜片钳技术体系, 也能独立服务于实验. 值得注意的是, 成像范围和成像质量呈负相关, 使用者需根据实验目的进行权衡选择. 进一步改良光路, 通过精准照明与限制无关区域的照明强度, 减弱光毒性, 实现了单通道记录区域的高质量单分子荧光成像, 且成像区域位置具备一定可调性. 将这一技术与细胞贴附式记录相结合, 构建可同步记录荧光信号和电信号的荧光膜片钳技术. 通过对mPiezo1进行单通道记录实验, 证实改良后的照明系统不会干扰细胞贴附式单通道记录. 本研究提出的技术方法, 为细胞膜上表面单分子荧光成像提供了简便有效的解决方案, 为未来开展蛋白质结构动力学与功能有关的研究提供了新的技术储备. 结合对目的蛋白的荧光标记, 未来研究者将能够依托此技术识别单通道记录区域中寡聚式通道蛋白的化学计量数或与蛋白功能相关的结构动力学信息. 对于一些潜在的以异源复合体形式发挥复杂功能的通道蛋白而言, 如黏附性G蛋白偶联受体LPHN2与跨膜通道样蛋白TMC1[36], 荧光膜片钳耦联技术能够通过单分子荧光信号反映二者的共定位或相互作用, 并以膜片钳技术同时记录其功能特征. 基于此技术方法, 开展蛋白结构动力学-功能解析工作, 可以进一步整合更多成熟的单分子荧光技术. 例如smFRET技术能够提供荧光标记位置点对点的亚纳米级结构动力学信息, 而queenFRET技术则能够帮助识别通道蛋白在膜法方向上的亚纳米级动力学变化, 这些技术整合将使结构-功能解析中的通道蛋白结构动力学信息拥有多维度的分析视角, 为后续研究和应用提供更为可靠的实验基础.
局域照明增强的活细胞单分子荧光-单通道膜片钳耦联技术
Local illumination-enhanced live-cell single-molecule fluorescence and single-channel patch-clamp coupling technology
-
摘要: 通道蛋白精确调控生命活动中物质跨膜转运, 为信号传递和能量代谢等复杂功能提供了结构保障. 单分子荧光技术与单通道膜片钳技术偶联对于解析其“结构-动力学-功能”的关联至关重要. 为解决二者联用中细胞内的高荧光背景限制单分子信号采集的难点, 本研究提出了一种选择性局部激发光路, 在活细胞上表面构建可控范围的局域照明场, 实现其中单分子荧光成像与动态追踪. 基于可调照明范围和区域, 达成照明光斑与玻璃电极的亚微米级共定位, 有效获取细胞贴附式单通道电流记录, 及高信噪比的单分子荧光时间轨迹. 本工作建立了一个可用于揭示通道蛋白结构-功能耦联机制的、具有普适性的单分子水平研究框架.Abstract: Channel proteins act as precise molecular regulators of transmembrane transport, which is a fundamental process essential for maintaining cellular homeostasis. These proteins dynamically modulate their functional states through conformational changes, thereby forming the structural basis for complex physiological processes such as signal transduction and energy metabolism. Single-molecule fluorescence spectroscopy and single-channel patch-clamp electrophysiology represent two cornerstone techniques in modern biophysics: the former enables molecular-resolution analysis of structural dynamics, while the latter provides direct functional characterization of ion channel activity. Despite their complementary capabilities, integrating these techniques to simultaneously monitor protein conformational dynamics and functional states remains technically challenging, primarily due to the strong autofluorescence background inherent in single-molecule imaging in cellular environments. To address this limitation, we develop a spatially selective optical excitation system capable of localized illumination. By integrating tunable optical modules, we generate a dynamically adjustable excitation field on living cell membranes, achieving precise spatial registration between the excitation volume and the patch-clamp recording site. This system achieves submicron-scale alignment between the excitation zone and the micropipette contact area, enabling simultaneous electrophysiological recording and background-suppressed fluorescence detection within the clamped membrane domain. Experimental validation demonstrates that the systemcan perform single-molecule fluorescence imaging and trajectory analysis within a specified observation areas, with imaging resolution inversely related to the size of the illuminated region. Optimized optical design allows for precise excitation targeting while minimizing background illumination, thereby achieving high signal-to-noise ratio single-molecule imaging and significantly reducing photodamage. Integration with cell-attached patch-clamp configurations establishes a dual-modality platform for synchronized acquisition of single-molecule fluorescence images and single-channel recordings. The validation using mechanosensitive mPiezo1 channels confirms the system’s compatibility with single-channel recording, indicating that optical imaging induces no detectable interference to electrophysiological signal acquisition. This method overcomes longstanding challenges in the simultaneous application of single-molecule imaging and electrophysiological techniques in live-cell environments. It establishes a novel experimental framework for investigating the structure-function relationships of channel proteins and membrane-related molecular machines through spatially coordinated optoelectronic measurements on live-cell membranes, which has broad applicability in molecular biophysics and transmembrane transport mechanism research.
-

-
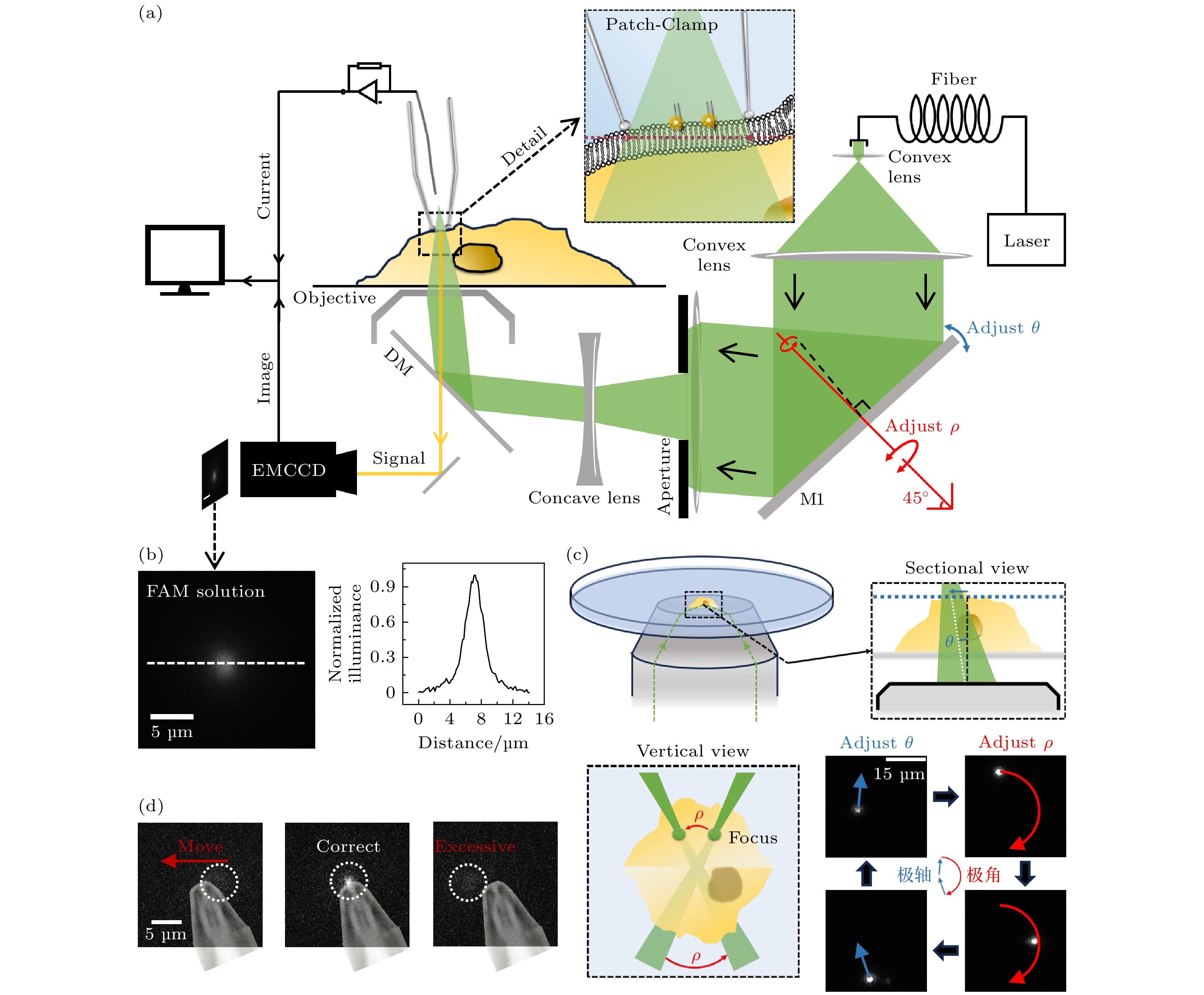
图 1 细胞膜上表面单分子荧光成像方法 (a) 细胞膜上表面单分子荧光成像光路图, 其中θ与ρ为光路中反射镜M1调整聚焦光斑的实际参数, M1沿着固定轴(红色轴)的旋转能够对ρ进行调整; (b) 在羧基荧光素(CF)溶液中单分子成像光斑图像以及白线截面的归一化强度分布; (c) 在羧基荧光素溶液中, 聚焦光斑位置通过反射镜M1的参数调节示意图与实际效果(右下), 其中虚线框图分别为侧剖面视图(右上)与正上方俯视图(左下); (d) 聚焦成像光斑通过调整位置实现与针尖匹配, 其中针尖图像为贴图标记, 白色虚线框指示了聚焦光斑位置
Figure 1. Cell membrane surface single-molecule fluorescence imaging technique: (a) Optical setup diagram for single-molecule fluorescence imaging on the upper leaflet of the cell membrane, in this diagram, θ and ρ represent the actual parameters adjusted by mirror M1 to focus the light spot, rotation of mirror M1 along the fixed axis (red-marked axis) enables precise control over the ρ parameter; (b) normalized intensity distribution of single-molecule imaging spot images and white reference line cross-section in carboxyfluorescein (CF) solution; (c) schematic diagram of focused laser spot position adjustment via mirror M1 parameters (bottom right) in CF solution, with experimental validation (bottom right). Dashed-line boxes denote: (top right) side cross-sectional view and (bottom left) top-down view; (d) alignment of the focused imaging laser spot with a probe tip via positional adjustment, where the tip image is overlaid as a reference marker, and white dashed boxes denote the focal spot location.
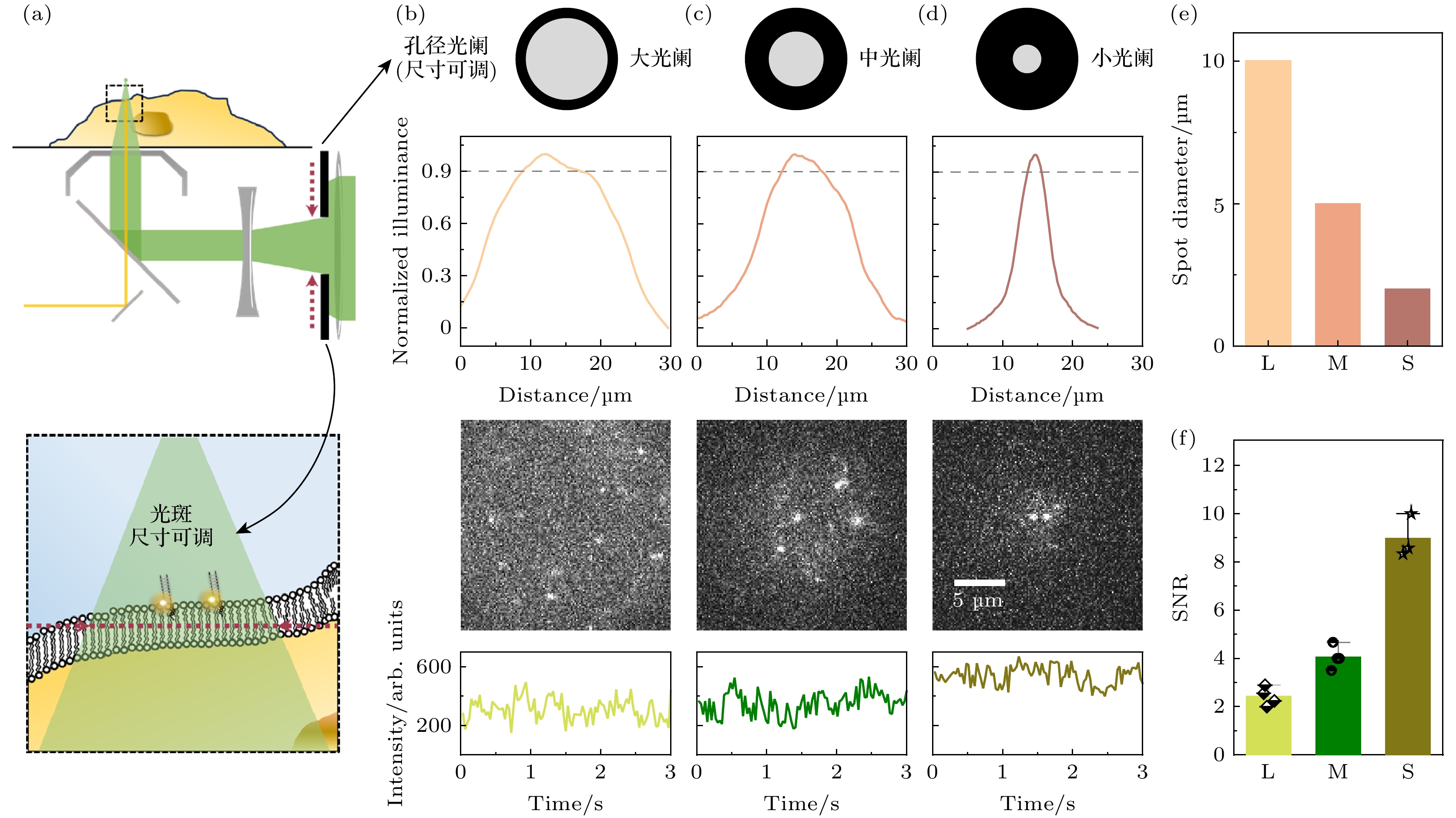
图 2 细胞膜上表面单分子荧光成像 (a) 扩大成像范围光路原理示意图, 上半图中黑色虚线框放大为下半图, 下半图中的红色虚线指代物镜的前焦面; (b)—(d) 可调光阑孔径变化可以影响成像光斑的尺寸和信噪水平; (e) 不同孔径的光阑所产生的成像光斑直径; (f) 不同孔径的光阑下单分子自由扩散信号的信噪比(SNR)
Figure 2. Single-molecule fluorescence imaging of the upper leaflet of the cell membrane: (a) Schematic diagram of expanded imaging field optical path design, the black dashed box in the upper panel is zoomed in as the lower panel, where the red dashed line indicates the front focal plane of the objective lens; (b)–(d) adjustment of the adjustable aperture diameter influences both the imaging spot size and the SNR; (e) variation in imaging spot diameter with aperture size; (f) SNR of single-molecule free diffusion signals under varying aperture diameters.
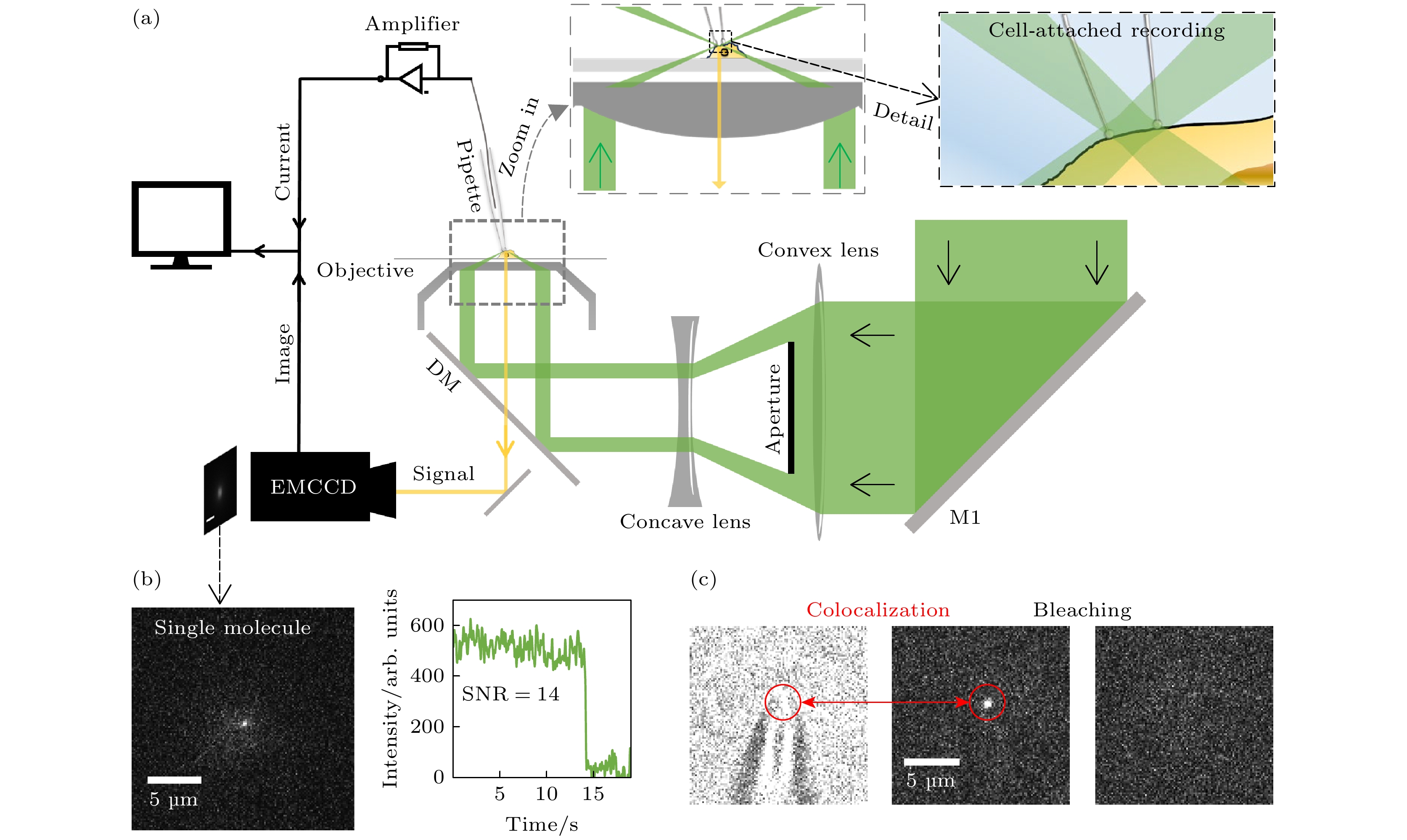
图 3 荧光膜片钳联用装置设置 (a) 荧光膜片钳联用装置示意图; (b) 在聚焦光斑处实现单分子荧光成像的实例及其数据展示; (c) 针尖与针尖荧光信号共定位以及针尖荧光信号的漂白
Figure 3. Fluorescence-patch-clamp system configuration: (a) Schematic diagram of fluorescence-patch-clamp system; (b) examples of single-molecule fluorescence imaging at focused laser spots and corresponding data; (c) colocalization of the probe tip and its fluorescence signal, along with photobleaching of the tip fluorescence.
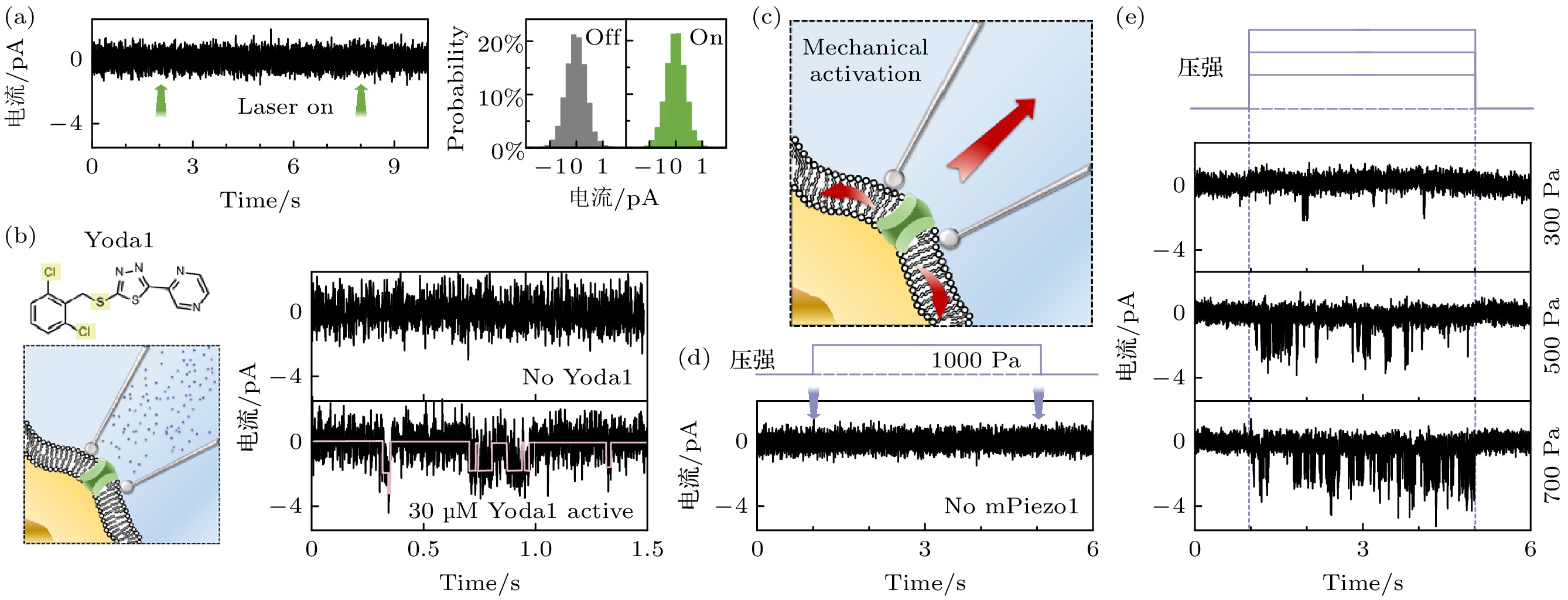
图 4 荧光膜片钳联用装置中膜片钳系统的性能 (a) 聚焦光斑对单通道记录不产生影响, 以及对应的统计图; (b) 使用Yoda1激活mPiezo1示意图以及mPiezo1的单通道信号; (c) 通过改变压强, 使细胞膜上产生机械应力以激活mPiezo1; (d) 在未转染mPiezo1的细胞上进行压力变化测试; (e) 在转染了mPiezo1的HEK293细胞上施加不同的压力变化以不同程度地激活钳制区域的mPiezo1
Figure 4. Performance evaluation of the patch-clamp system in fluorescence-patch-clamp system: (a) The focused laser spot has no measurable impact on single-channel electrophysiological recordings, as demonstrated by statistical analysis; (b) schematic of mpiezo1 activation using yoda1 and single channel signaling of mPiezo1; (c) by changing the pressure, mechanical stress is generated on the cell membrane to activate mPiezo1; (d) pressure change tests on cells not transfected with mPiezo1; (e) application of differential mechanical pressure to mPiezo1-transfected HEK293 cells to induce region-specific activation of mPiezo1.
-
[1] Jiang D, Banh R, El-Din T M G, Tonggu L, Lenaeus M J, Pomes R, Zheng N, Catterall W A 2021 Cell 184 5151 doi: 10.1016/j.cell.2021.08.021 [2] Yang X Z, Lin C, Chen X D, Li S Q, Li X M, Xiao B L 2022 Nature 604 377 doi: 10.1038/s41586-022-04574-8 [3] Jentsch T J, Pusch M 2018 Physiol. Rev. 98 1493 doi: 10.1152/physrev.00047.2017 [4] Cui Y N, Yu M, Yao X M, Xing J J, Lin J X, Li X J 2018 Mol. Plant 11 1315 doi: 10.1016/j.molp.2018.09.008 [5] Weiss S 2000 Nat. Struct. Biol. 7 724 doi: 10.1038/78941 [6] Smiley R D, Hammes G G 2006 Chem. Rev. 106 3080 doi: 10.1021/cr0502955 [7] Drapeau P, Ali D W, Buss R R, Saint-Amant L 1999 J. Neurosci. Methods 88 1 doi: 10.1016/S0165-0270(99)00008-4 [8] Neher E, Sakmann B 1976 Nature 260 799 doi: 10.1038/260799a0 [9] Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J 1981 Pflug. Arch. Euro. J. Phys. 391 85 doi: 10.1007/BF00656997 [10] Mann S A, Heide J, Knott T, Airini R, Epureanu F B, Deftu A F, Deftu A T, Radu B M, Amuzescu B 2019 J. Pharmacol. Toxicol. Methods 100 1065990 doi: 10.1016/j.vascn.2019.106599 [11] Mason M J, Simpson A K, Mahaut-Smith M P, Robinson H P C 2005 Biophys. J. 88 739 doi: 10.1529/biophysj.104.049866 [12] Alcami P, Franconville R, Llano I, Marty A 2012 J. Neurosci. 32 3118 doi: 10.1523/JNEUROSCI.5371-11.2012 [13] Maki B A, Cummings K A, Paganelli M A, Murthy S E, Popescu G K 2014 J Vis Exp. 88 e51629 doi: 10.3791/51629 [14] Ludewig U, Pusch M, Jentsch T J 1997 Biophys. J. 73 789 doi: 10.1016/S0006-3495(97)78111-6 [15] Vazetdinova A, Valiullina-Rakhmatullina F, Rozov A, Evstifeev A, Khazipov R, Nasretdinov A 2022 Front. Mol. Neurosci. 15 979479 doi: 10.3389/fnmol.2022.979479 [16] Bykova E A, Zhang X D, Chen T Y, Zheng J 2006 Nat. Struct. Mol. Biol. 13 1115 doi: 10.1038/nsmb1176 [17] Alford R, Ogawa M, Hassan M, Gandjbakhche A H, Choyke P L, Kobayashi H 2010 Contrast Media Mol. Imaging 5 1 doi: 10.1002/cmmi.360 [18] Yildiz A, Forkey J N, McKinney S A, Ha T, Goldman Y E, Selvin P R 2003 Science 300 2061 doi: 10.1126/science.1084398 [19] Gill J K, Shaw G S 2024 Chembiochem 25 e202400193 doi: 10.1002/cbic.202400193 [20] 贾棋, 樊秦凯, 侯文清, 杨晨光, 王利邦, 王浩, 徐春华, 李明, 陆颖 2021 物理学报 70 158701 doi: 10.7498/aps.70.20210707 Jia Q, Fan Q K, Hou W Q, Yang C G, Wang L B, Wang H, Xu C H, Li M, Lu Y 2021 Acta Phys. Sin. 70 158701 doi: 10.7498/aps.70.20210707 [21] 陈泽, 马建兵, 黄星榞, 贾棋, 徐春华, 张慧东, 陆颖 2018 物理学报 67 118201 doi: 10.7498/aps.67.20180501 Chen Z, Ma J B, Huang X Y, Jia Q, Xu C H, Zhang H D, Lu Y 2018 Acta Phys. Sin. 67 118201 doi: 10.7498/aps.67.20180501 [22] Li Y, Qian Z Y, Ma L, Hu S X, Nong D G, Xu C H, Ye F F, Lu Y, Wei G H, Li M 2016 Nat. Commun. 7 12906 doi: 10.1038/ncomms12906 [23] 樊秦凯, 杨晨光, 胡书新, 徐春华, 李明, 陆颖 2023 物理学报 72 147801 doi: 10.7498/aps.72.20230450 Fan Q K, Yang C G, Hu S X, Xu C H, Li M, Lu Y 2023 Acta Phys. Sin. 72 147801 doi: 10.7498/aps.72.20230450 [24] Hou W Q, Ma D F, He X L, Han W J, Ma J B, Wang H, Xu C H, Xie R P, Fan Q H, Ye F F, Hu S X, Li M, Lu Y 2021 Nano Lett. 21 485 doi: 10.1021/acs.nanolett.0c03941 [25] 马东飞, 侯文清, 徐春华, 赵春雨, 马建兵, 黄星榞, 贾棋, 马璐, 刘聪, 李明, 陆颖 2020 物理学报 69 038701 doi: 10.7498/aps.69.20191607 Ma D F, Hou W Q, Xu C H, Zhao C Y, Ma J B, Huang X Y, Jia Q, Ma L, Liu C, Li M, Lu Y 2020 Acta Phys. Sin. 69 038701 doi: 10.7498/aps.69.20191607 [26] Jiang D H, Tonggu L G, Gamal El-Din T M, Banh R, Pomès R, Zheng N, Catterall W A 2021 Nat. Commun. 12 128 doi: 10.1038/s41467-020-20078-3 [27] Levring J, Terry D S, Kilic Z, Fitzgerald G, Blanchard S, Chen J 2023 Nature 616 606 doi: 10.1038/s41586-023-05854-7 [28] Tokunaga M, Imamoto N, Sakata-Sogawa K 2008 Nature Methods 5 159 doi: 10.1038/nmeth1171 [29] Sasmal D K, Yadav R, Lu H P 2016 J. Am. Chem. Soc. 138 8789 doi: 10.1021/jacs.6b03496 [30] Sasmal D K, Lu H P 2014 J. Am. Chem. Soc. 136 12998 doi: 10.1021/ja506231j [31] Hilgemann D W, Lu C C 1998 Ion Channels, Pt B 293 267 doi: 10.1016/S0076-6879(98)93018-X [32] Couey J J, Ryan D P, Glover J T, Dreixler J C, Young J B, Houamed K M 2002 Neurosci. Lett. 329 17 doi: 10.1016/S0304-3940(02)00556-6 [33] Lee C H, Wallace D C, Burke P J 2024 Mitochondrial communications 2 38 doi: 10.1016/j.mitoco.2024.03.001 [34] Icha J, Weber M, Waters J C, Norden C 2017 Bioessays 39 1700003 doi: 10.1002/bies.201700003 [35] Syeda R, Xu J, Dubin A E, Coste B, Mathur J, Truc H, Matzen J, Lao J, Tully D C, Engels I H, Petrassi H M, Schumacher A M, Montal M, Bandell M, Patapoutian A 2015 Elife 4 e07369 doi: 10.7554/eLife.07369 [36] Yang Z, Zhou S H, Zhang Q Y, Song Z C, Liu W W, Sun Y, Wang M W, Fu X L, Zhu K K, Guan Y, Qi J Y, Wang X H, Sun Y N, Lu Y, Ping Y Q, Xi Y T, Teng Z X, Xu L, Xiao P, Xu Z G, Xiong W, Qin W, Yang W, Yi F, Chai R J, Yu X, Sun J P 2025 Cell Res. 35 243 doi: 10.1038/s41422-025-01075-x -
计量
- 文章访问数: 119
- HTML全文浏览数: 119
- PDF下载数: 3
- 施引文献: 0