
 首页
首页 登录
登录 注册
注册



-
在微电子工业中, 硅化学气相沉积(chemical vapor deposition, CVD)是制备硅薄膜的常规方法, 在此过程中硅烷会分解成为各种自由基如Si, SiH以及
$ {\text{SiH}}_n^ + $ 阳离子等. 对于这些自由基的光谱学研究能够为CVD诊断技术提供一定的理论依据, 因此, 近年来含硅自由基越来越受到材料科学和理论化学研究工作者的重视, 围绕着这些分子离子的研究层出不穷[1–6]. 毫无疑问, 在这些自由基当中, SiH2是最重要的中间产物之一, 因此对于SiH2光谱及相关反应的研究一直备受关注. 在实验方面, Dubois等[7,8] 从1967—1968年间首次测量了SiH2的吸收谱. 1986年, Thoman和Steinfeld[9]报道了SiH2自由基的荧光寿命. 4年之后, Ishikawa等[10]通过激光诱导荧光和受激辐射泵入技术观察到该自由基的高激发态. 1999年, Hirota 与Ishikawa[11]合作报道了低振动能级的高精度谱, 并且给出了非常精确的分子常量.理论方面, 主要包含势能面(potential energy surface, PES)的构建和动力学研究两部分. 2004年, Yurchenko等[12]构建了SiH2两个最低电子态的从头计算势能面并且计算了振转能量. 2006年, Tokue等[13,14]构建了该体系最低的3个电子态的插值从头计算势能面, 发现了该体系不同电子态之间的锥形交叉, 并且研究了其光离解过程. 2017年, Wu等[15]采用高水平从头计算法系统地研究了Si+H2反应, 证实了该体系的锥形交叉(conical intersection, CI)结构, 研究了该反应中的几何相(geometric phase, GP)效应.
2021年, Cao等[16]采用置换不变多项式神经网络(permutational invariant polynomial neural network, PIP-NN)方法构建了一个SiH2基态全维势能面, 且基于该势能面研究了Si+H2→H+SiH反应的刚性聚合物(ring polymer molecular dynamics, RPMD)分子反应动力学及其同位素效应. 1年后, Wang 等[17]基于8953个从头计算能量点, 用包含Davidson修正的多参考组态相互作用(multi-reference configuration interaction MRCI)方法构建了SiH2基态多体展开势能面, 该势能面的总方均根偏差为0.0085 eV, 势能面的稳定点与实验值和其他理论值吻合得非常好. 基于该势能面, 该团队运用含时波包(time-dependent wave packet, TDWP)方法, 计算了
$ {\text{Si}}{(^1}{\text{D}}) + {{\text{H}}_2}({{\text{X}}^1}\Sigma _g^ + ) \to {\text{H}}{(^2}{\text{S}}) + {\text{SiH}}({{\text{X}}^2}\Pi ) $ 反应的积分散射截面和速率常数, 与Cao等[16]的理论计算结果符合得很好.三原子体系由于具有较少的自由度, 可以实现非含时理论方法的精确计算, 如ABC计算程序[18], 也可以采用近年来应用比较普遍的TDWP方法[19,20]. 本工作正是基于Wang 等[17]所构建的
$ {\text{Si}}{{\text{H}}_2}({1^1}{\text{A}}') $ 势能面, 用切比雪夫量子波包方法[21–23]对$ {\text{H}}{(^2}{\text{S}}) + {\text{SiH}}({{\text{X}}^2}\Pi ) \to {\text{Si}}{(^1}{\text{D}}) + {{\text{H}}_2}({{\text{X}}^1}\Sigma _g^ + ) $ 反应进行动力学研究. 这种方法的优越性在于只需要波包的一次传播就能够给出所有能量点处的反应概率, 波包在实空间的传播没有近似, 因而在理论计算中具有优势[24,25]. 本文结构如下: 第2节介绍所采用的理论方法以及相应的数值计算方法; 第3节是理论计算结果和讨论. 第4节总结相关的结论. 除特别说明之外, 文中的所有公式和计算都使用原子单位. -
在雅可比坐标下, 反应物H +SiH体系的哈密顿量的表达式为
其中R 表示H与SiH质心间的距离, r表示SiH 内部原子间距,
$ {\mu _{\text{r}}}{ = }{{{m_{{\text{Si}}}}{m_{\text{H}}}} {/ } {({m_{{\text{Si}}}} + {m_{\text{H}}})}} $ 表示SiH双原子约化质量,$ {\mu }_{\text{R}} = {m}_{\text{H}}\left({m}_{\text{Si}}+{m}_{\text{H}}\right)/(2{m}_{\text{H}}+{m}_{\text{Si}}) $ 表示H + SiH三体约化质量, V(R, r, γ) 为$ {\text{Si}}{{\text{H}}_2} ({1^1}{\text{A}}') $ 体系的势能.$ \hat j $ 和$ \hat l $ 是双原子转动角动量算符和轨道角动量算符.$ {\hat l^2} $ 可以表示为(2)式中
$ \hat J $ 为总角动量算符,$ {\hat J_ + } $ ($ {\hat J_ - } $ ) 和$ {\hat j_ + } $ ($ {\hat j_ - } $ ) 表示总角动量升(降)算符和转动角动量升(降)算符,$ {\hat J_z} $ 和$ {\hat j_z} $ 分别是$ \hat J $ 和$ \hat j $ 在定体(body fixed, BF)坐标系中z轴的投影. 为了简单起见, 令BF 坐标系z轴与R的方向一致, 即SiH质心 指向另一个H的方向. (2)式等号右侧最后两项对应于科里奥利耦合(Coriolis coupling, CC)效应.传播得到的波包利用下面所给出的递推关系, 可以避免离散误差和插值误差的引入[26,27]:
其中,
$ {\hat H_{{\text{norm}}}} = {{( {\hat H - {H^ + }} )} {/ } {{H^ - }}} $ 为归一化哈密顿量,$ {H^ \pm } = {{\left( {{H_{\max }} \pm {H_{\min }}} \right)} {/ } 2} $ , 而$ {H_{\max }} $ ($ {H_{\min }} $ )通过光谱的边界条件计算获得[28]. 在格点边缘, 用高斯型衰减函数D来描述输出波的边界条件:$ {d_x} $ 控制阻尼范围,$ {x_{\text{d}}} $ 为阻尼起点. 初态波包$ \left| {{\psi _0}} \right\rangle $ 为式中
$ N $ ,$ {k_0} $ ,$ {R_0} $ 和$ \delta $ 分别表示归一化常量, 初态波包的动量、中心位置和宽度,$ \left| {{\varphi _{\text{i}}}} \right\rangle $ 一个具体的振转本征函数.总反应概率可以在产物通道的分界面通过反应流算法[29]获得, 这种分界面在产物渐近线上并不要求S矩阵元完全收敛, 因此, 流算法可以很方便地应用于反应物雅可比坐标系中. 应用波包的传播, 反应概率可以表示为
式中
$ \theta \equiv \arccos \left[ {{{\left( {E - {H^ + }} \right)} {/ } {{H^ - }}}} \right] $ 是切比雪夫角,$ r = {r_{\text{f}}} $ 处为产物通道的分界面,$ {a_{\text{i}}}(E) $ 为初始波包的能量振幅[30], 表示为其中,
$ {{\text{h}}_{{\text{li}}}}^{(2)}({k_{\text{i}}}R) $ 表示第二类汉克尔函数.对所有J的反应概率进行求和, 得到给定初态
$ \left( {{\upsilon _{\text{i}}}{j_{\text{i}}}{l_{\text{i}}}} \right) $ 的积分散射截面(integral cross section, ICS)其中
$ {E_{\text{c}}} $ 表示碰撞能量,$ k_{{\upsilon _{\text{i}}}{j_{\text{i}}}}^2 = 2{\mu _{\text{R}}}{E_{\text{c}}} $ , 角标p表示宇称. 将积分散射截面$ {\sigma _{{\upsilon _{\text{i}}}{j_{\text{i}}}}}({E_{\text{c}}}) $ 对碰撞能量$ {E_{\text{c}}} $ 结合玻尔兹曼权重进行积分, 得到反应速率常数为式中f, kB和T分别是电子的简并因子、玻尔兹曼常量和热力学温度.
-
动力学计算所采用的SiH2(11A′)势能面由文献[17]提供, 图1描述了该势能面的拓扑性质. 从图中能看出PES中主要的稳定点: 全局最小(global minimum, GM)点, 过渡态(transition-state, TS)以及鞍点(saddle point, SP). 图1(a)为γ = 91.5°时的势能曲线, 横坐标为H与SiH质心间的距离, 纵坐标为SiH之间的距离, 图中可以很清楚地看到GM点. 相对于产物渐近线来说, 全局最小点的能量为–2.66 eV, 位于RSiH = RSiH = 2.866a0, 这个结果与实验值RSiH = 2.87a0, ∠HSiH = 92.1°[8]吻合得非常好, 与稍早的理论计算结果[16]全局最小点能量–2.63 eV, RSiH = 2.90a0, ∠HSiH = 92.5°也非常符合. 图1(b)为T构型势能曲线, 即Si原子垂直于H-H键垂直插入H-H中心的情况, 横坐标表示Si与H-H质心间距, 纵坐标表示H-H间距离, 这个图描写了一个C2v的对称结构, 从其中能观察到GM点以及一个TS. 图1(c)为H-Si-H直线构型下的势能曲线, 图中在RSiH = 2.736a0存在一个过渡态, 其能量高于GM 点2.70 eV. 图1(d)为直线构型Si-H-H下的势能曲线, 该构型下在RHH = 2.358a0 , RSiH = 5.407a0处存在一个鞍点, 其能量在产物Si+H2渐近线之上1.01 eV. 这些图中势能曲线描绘的所有区域都是光滑的, 无任何拐点, 说明这是一个高质量的势能面, 适合于用来进行相关的动力学研究.
图2描述H原子从30°, 60°, 90°, 120°, 150°和180°插入SiH时, SiH2体系的势能随着反应坐标
$ {R_{{\text{SiH}}}} - {R_{{\text{HH}}}} $ 的变化关系, 即最小能量路径(minimum energy path, MEP). 很显然$ {\text{H}} + {\text{SiH}} \to {\text{Si}} + {{\text{H}}_2} $ 是一个放热反应, 产物渐近线与反应物渐近线的能量差为1.16 eV, 这与文献[17]报道的其反方向反应吸热0.81 eV存在较大的差异. 而在横坐标$ {R_{{\text{SiH}}}} - {R_{{\text{HH}}'}} $ 为6a0—8a0之间存在一个小平台, 所有MEP曲线都在这里交合, 这个平台到产物渐近线的能量差恰好为0.81 eV. 观察MEP, 在30°时, 存在一个3.44 eV的深势阱, 随着角度的增大, 势阱的深度逐渐变小. MEP表明在最小能量路径中跟随着深势阱之后存在着一个鞍点, 因此图2中可以看到30°, 150°, 180°角对应的MEP在产物通道附近存在着小势垒结构, 高度最大的一个势垒相对于Si+H2产物渐近线为1.04 eV, 这些势垒将会阻碍H2的形成. -
基于上述势能面, 本工作采用波包传播方法研究
$ \text{H}({}^{2}\text{S})+\text{SiH}({\text{X}}^{2}\Pi )\to $ $ \text{Si}({}^{1}\text{D})+{\text{H}}_{2}({\text{X}}^{1}{\Sigma }_{\text{g}}^{+}) $ 反应动力学. 结合势能面中的深势阱以及SiH2体系的大质量, 计算中需要高的碰撞能量、大量的分波和基组数, 这给动力学计算带来了极大的挑战, 尤其是在CC计算中, 需要耗费大量的计算资源. 对于$ J = 0 $ 的情况, 经过广泛的收敛性测试, 选取的最优的计算参数列于表1. 应用这些计算参数, 对于J = 0, 5, 15, ···, 90的分波进行了耦合态(coupled-state, CS)近似和包含CC效应的精确量子力学计算, 其他反应概率通过上述反应概率用3次样条插值法插值得出[31], 进而算出碰撞能量范围在0.001—1.0 eV 的积ICS, 最后给出速率常数. -
图3所示为CS和CC两种计算方法下对于给定初始状态的H+SiH(ν = 0, j = 0)反应在角动量量子数J = 0, 10, 20, 30, 40, 50, 60, 70的反应概率. 由J = 0的概率曲线可以看出, 该反应为无势垒的放热反应. 随着总角动量J的增大, 反应的阈能逐渐增大, 这是由于离心势垒增大引起的. 比较两种曲线可以看出CS和CC概率的差异主要表现在3个方面. 第一, CC概率振荡幅度和频率都比CS曲线小, 这是由于深势阱中的络合物机制导致, H靠近SiH的过程中囿于深势阱中形成络合物SiH2, 造成了反应概率尤其是CS概率中密集的振荡结构, 而CC概率的振荡频率与幅度都小是因为反应中科里奥利耦合受到长程力支配的原因. 第二, 随着碰撞能量的增大, 概率的变化比较复杂, 当J ≤ 10 (J ≤ 40)时, CS(CC)概率随着碰撞能量增大而减小, 当J ≥ 20 (J ≥ 50)时, CS(CC)概率随着碰撞能量增大呈现先增大后减小的趋势. 第三, 随着J的增大, CS与CC反应概率都会减小. 当J ≤ 30, CC与CS概率差别比较小, J ≥ 40时, CC概率明显大于CS的值例如, 当J = 70时, CS概率基本为零, 从放大的插图中可以观察到, 在碰撞能量为0.99 eV时, CC概率是CS概率的70倍. 这是因为当J增大到一定程度时, CC计算中用到更多的分波, 通过CC效应破坏中间络合物促进产物的生成. 所以, 随着J的增大, CS与CC概率的差别越来越明显.
-
根据(8)式, 积分散射截面是所有分波反应概率的加权求和, 在一定的能量下, 当J 较小时, 因为简并因子2J+1, 分波贡献最初随着J的增大而增大, 然后当J继续增大时, 因为反应阈向高能区移动, 分波贡献随着J 的增大呈现断崖式衰减, 如图4所示. 图4(a), (b)分别为两种计算方法所得的分波贡献, 显然,能量越高, 所需的分波越多. 在CS和CC模型中, 需要计算分波的最大角动量量子数分别为J = 80和J = 90. 对比4(a), (b)发现, 在相同的能量下, CC对应的分波贡献远大于CS模型, 这与反应概率的表现是一致的.
图5描述了H+SiH反应在
$ 1.0 \times {10^{ - 3}} $ —1.0 eV的碰撞能量范围下ICS随碰撞能量的变化, 与反应概率表现不同的是ICS曲线中没有密集的振荡, 尤其是CC反应截面曲线几乎是光滑的, 其原因是ICS计算中不同角动量反应概率加权平均的效应. 曲线表明, ICS随着能量的增大单调递减, 在低能区域内随着碰撞能量的增大急剧减小, 高能区随着能量的增大, ICS缓慢减小. 这些动力学行为都符合无势垒的放热反应机制. 比较两种计算结果发现, 忽略CC效应使得ICS减小, CS与CC ICS的差距在低能区域尤其明显. CC方法计算所得的ICS, 无论是振荡频率还是幅度都比CS模型小很多, 曲线比较光滑, 基本没有共振结构. 这与计算方法有关系, 在CC计算中用到更多的分波, 加权求和计算ICS导致绝大部分共振被消除. 此外,与相似反应H+CH[32]相比较发现, 在整个能量范围内, H+SiH反应ICS都大于H+CH, 例如在碰撞能量为0.01 eV时, H+SiH反应ICS值为60.08 Å2, 而H+CH反应ICS值为10.01 Å2, 二者大小相差悬殊. 这是由于势能面的特点决定的, 比如, SiH2的全局最小点相对Si+H2渐近线为–2.66 eV, CH2的全局最小点相对C+H2渐近线为–8.22 eV, 又如, 在MEP中, ∠HHSi或∠HHC = 180°时, 势阱相对于产物渐近线势垒高度分别为1.35 eV和1.06 eV. 势能面的这些特点对上述动力学结果有决定性的影响. -
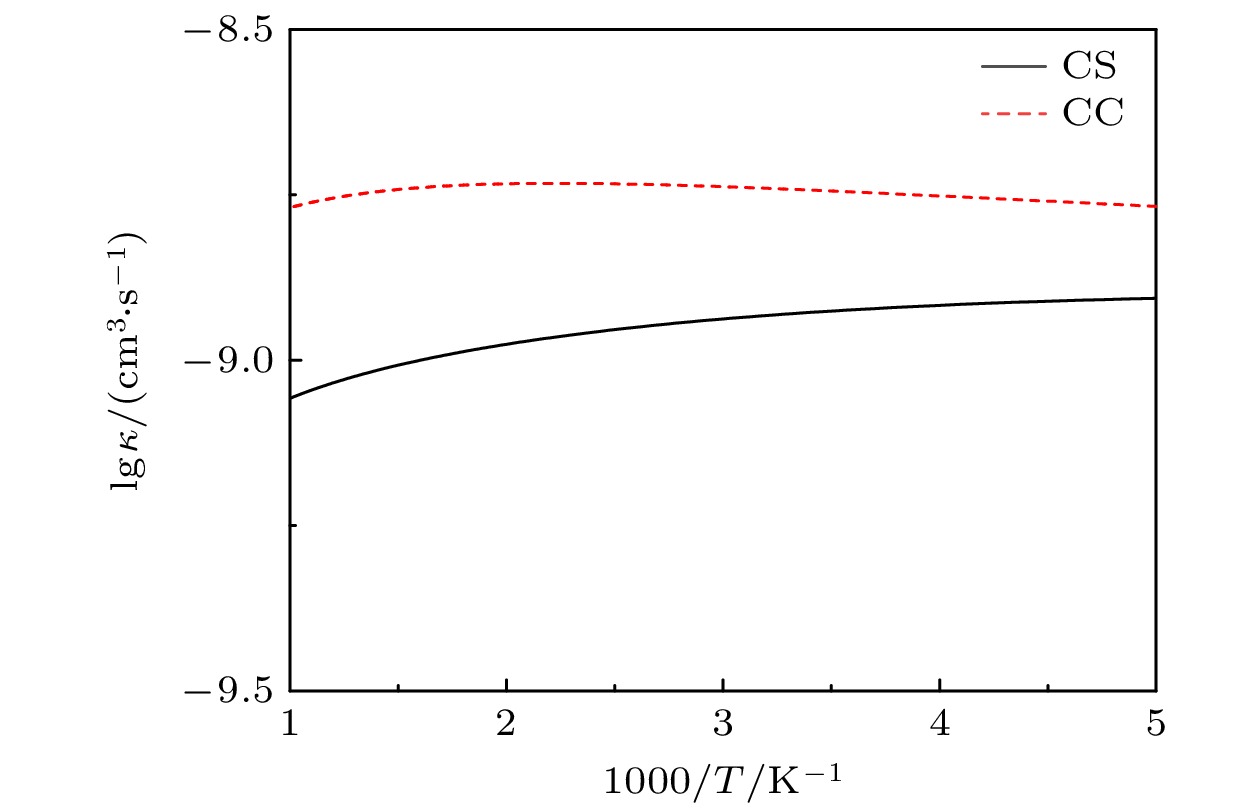
图6为温度在200—1000 K之间速率常数的阿伦尼乌斯(Arrhenius)图. 很显然, 在整个温度范围内CC速率都大于CS速率, 根据(9)式, 速率常数是ICS的加权积分结果, 对比图5所示的该反应CC和CS的ICS , 这样的结果是并不奇怪. 由图6还可以看出, CC速率随温度变化很小, CS速率则随温度升高而减小. 因此, 随着温度的升高, CS速率与CC速率的差值在变大, 例如, 在T = 200 K时, 两者差值为4.6×10–10 cm3/(molecule·s), 在T = 400 K时, 这个差值变成为7.3×10–10 cm3/(molecule·s). 可见, 忽略CC效应使H+ SiH反应的速率常数显著减小. 另外, H+ SiH反应CC速率与H+ CH[33]反应相比较, 非常相似的一点是在300—1000 K之间两个反应的速率几乎都是常数, 从数值上比较, H+ SiH反应速率常数为1.8×10–9 cm3/(molecule·s), H+ CH反应速率常数为1.01×10–10 cm3/(molecule·s), 前者是后者的18倍.
-
本文基于2022年构建的一个全维高精度势能面, 应用波包传播方法揭示了放热反应H+ SiH的动力学行为机制. 在不超过1.0 eV的碰撞能量下, 用CS近似和精确的CC方法得到了该反应的ICS和速率常数. 计算结果表明忽略科里奥利耦合效应会使H+ SiH反应的积分散射截面和速率常数大幅减小, 说明科里奥利耦合效应对该反应的影响非常强烈. 与H+CH反应相比较, 两个反应的ICS和速率常数随着能量变化的行为比较类似, 但在数值上也存在显著的区别. 这些动力学行为主要取决于势能面中的深势阱. 基于本工作所采用势能面的精确性, 期待本文H+ SiH动力学的研究能为该体系未来的实验或者理论研究提供一定的参考.
虽然本工作主要研究的是H+SiH绝热反应动力学, 但是目前团队也意识到了研究非绝热反应也是必要的, 特别是CI以及与之相关的GP效应[34–36], 所以基于现有的势能面来研究该体系的非绝热反应动力学可能是一项有意义的工作.
-
本篇论文的关联数据可以在科学数据银行
https://doi.org/10.57760/sciencedb.j00213.00052 中获取.
基于一个新SiH2(11A′)势能面的H+SiH反应动力学研究
Quantum dynamics study of reaction H+SiH using a new potential energy surface of SiH2(11A′)
-
摘要: 本文基于2022年报道的一个SiH2(11A′)势能面, 运用切比雪夫波包方法对$ \text{H}{(}^{2}\text{S})+\text{SiH}({\text{X}}^{2}\Pi ; \nu = 0, $$ j = 0)\to \text{Si}{(}^{1}\text{D})+{\text{H}}_{2}({\text{X}}^{1} \Sigma_{g}^{+}) $反应体系在$ 1.0 \times {10^{ - 3}} $—1.0 eV的碰撞能量范围内进行动力学研究. 分别应用忽略科里奥利耦合效应的耦合态近似和精确量子力学计算得到该反应的反应概率、积分散射截面和速率 常数. 计算发现在J 较大时, 科里奥利耦合效应显著提升该反应的反应概率, 忽略科里奥利耦合效应会使H + SiH 反应的积分散射截面和速率常数减小, 对于速率常数而言, 温度越高, 两种计算方法所得结果的差距越大. 精确的量子力学计算结果表明, H + SiH 反应的速率常数在300—1000 K之间几乎不随温度改变, 这与H + CH 反应非常相似, 但是在数值上, 前者比后者大1个数量级.Abstract: Initial state-selected and energy-resolved reaction probabilities, integral cross sections(ICSs), and thermal rate constants of the $ \text{H}{(}^{2}\text{S})+S\text{iH}({\text{X}}^{2}\Pi; \nu = 0\text{ },j = 0)\to \text{Si}{(}^{1}\text{D})+{\text{H}}_{2}({\text{X}}^{1} \Sigma_{g}^{+}) $ reaction are calculated within the coupled state(CS) approximation and accurate calculation with full Coriolis coupling(CC) by a time-dependent wave packet propagation method (Chebyshev wave packet method). Therefore, a new ab initio global potential energy surface (PES) of the electronic ground state (11A′) of the system, which was recently reported by Li et al. [
Phys. Chem. Chem. Phys. 2022 24 7759 ], is employed. The contributions of all partial waves to the total angular momentum J = 80 for CS approximation and J = 90 for CC calculation are considered to obtain the converged ICSs in a collision energy range of 1.0 ×10–3-1.0 eV. The calculated probabilities and ICSs display a decreasing trend with the increase of the collision energy and show an oscillatory structure due to the SiH2 well on the reaction path. The neglect of CC effect will lead to underestimation of the ICS and the rate constant due to the formation of an SiH2 complex supported by the stationary points of the SiH2(11A′) PES. In addition, the results of the exact calculation including CC effect are compared with those calculated in the CS approximation. For the reaction probability, CC and CS calculations change with similar tends, shown by their observations at small total angular momentum J = 10, 20 and 30, and the CC results are larger than the CS results almost in the whole considered energy range at large total angular momentum J = 40, 50, 60 and 70. The gap between CS and CC probability get more pronounced with increasing of J, which reveals that Coriolis coupling effects become more and more important with J increasing for the title reaction. Moreover, the exact quantum-wave calculations show that the thermal rate constant between 300 K and 1000 K for the title reaction shows a similar temperature independent behavior to that for the H + CH reaction, but the value of the rate constant for the H + SiH reaction is an order of magnitude larger than that for the H + CH reaction.-
Key words:
- reaction probability /
- integral cross section /
- rate constant .
-

-
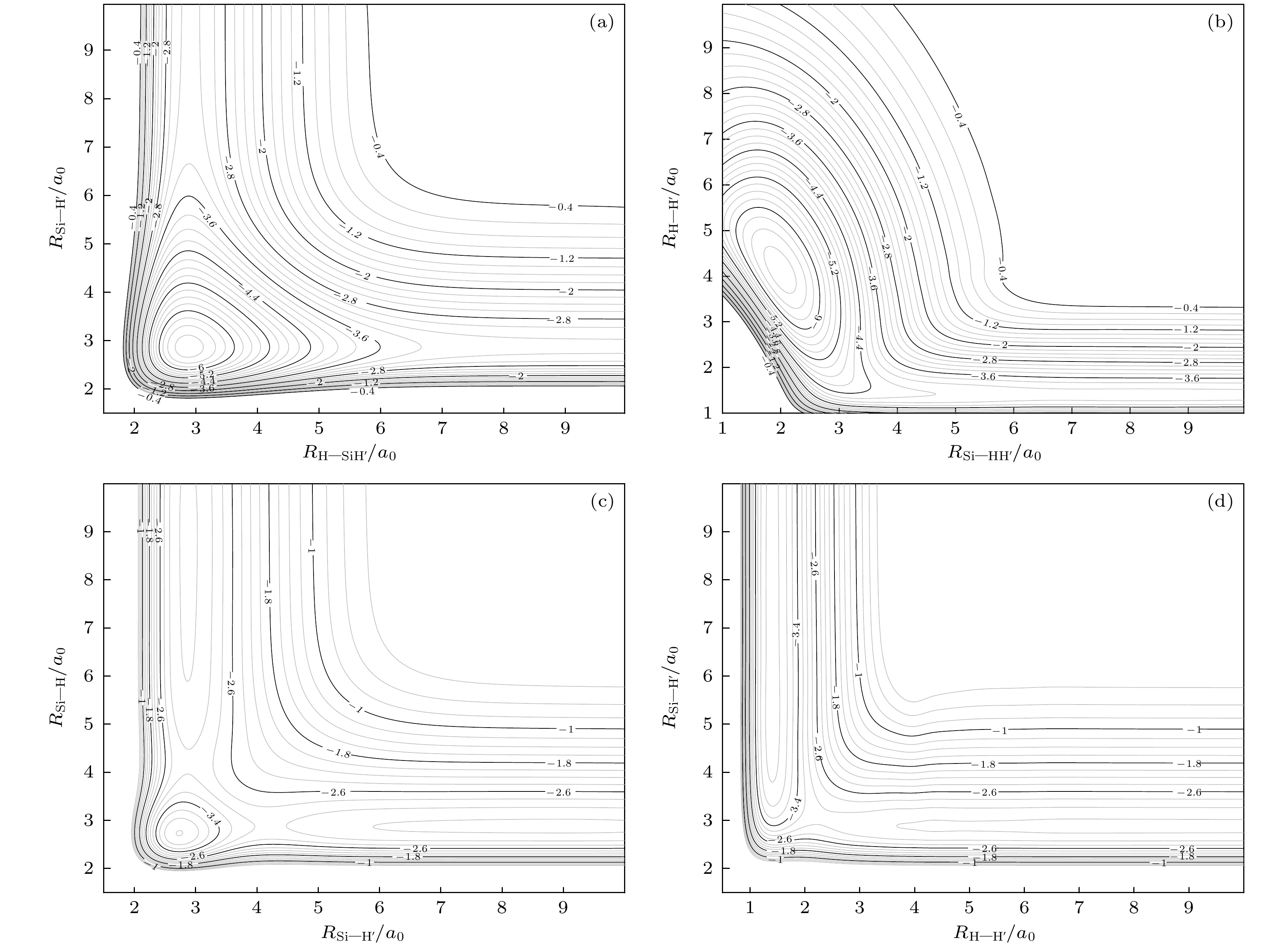
图 1
$ {\text{Si}}{{\text{H}}_2} $ 等势线, 图中等势线间隔为0.2 eV (a)γ = 91.5°, 以雅可比坐标RH-SiH和RSi-H为横纵坐标; (b) γ = 90°, 以雅可比坐标RSi–HH和RHH为横纵坐标; (c) H-Si-H线性结构, 以RSi–H和RSi–H为横纵坐标; (d) Si-H-H线性结构, 以RH–H和RSi–H为横纵坐标; 4个图的势能线起始点分别是–6.713, –6.713, –4.008, –4.084 eV, 等高线增量为0.1 eVFigure 1. Equipotential contour plot for SiH2: (a) Contour plot for bond stretching as a function of the product Jacobi coordinates RH–SiH and RSi–H with the Jacobi angle γ = 91.5°; (b) as a function of the reactant Jacobi coordinates RSi—HH and RHH with the Jacobi angle γ = 90°; (c) linear H-Si-H geometry, using RSi–H and RSi–H as the horizontal and vertical coordinates; (d) linear Si-H-H geometry, using RH–H and RSi–H as the horizontal and vertical coordinates; the contour increments are 0.1 eV, and the four panels starting from –6.713, –6.713, –4.008, –4.084 eV.
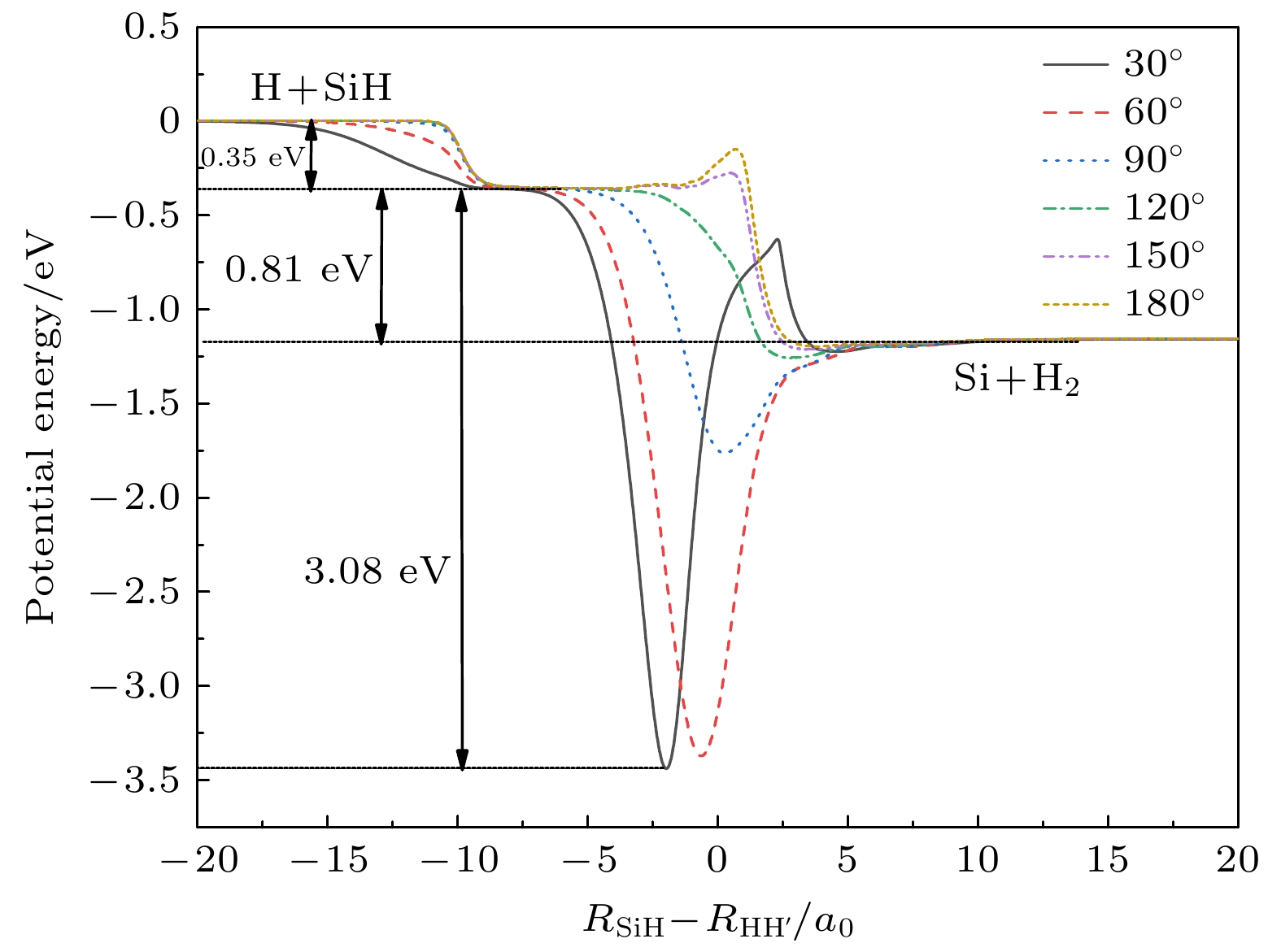
图 2 最小能量路径, ∠[H-H-Si]分别为30°, 60°, 90°, 120°, 150°, 180°
Figure 2. The MEP of different approaching angles, ∠[H-H-Si] = 30°, 60°, 90°, 120°, 150°, 180° for the title reaction.
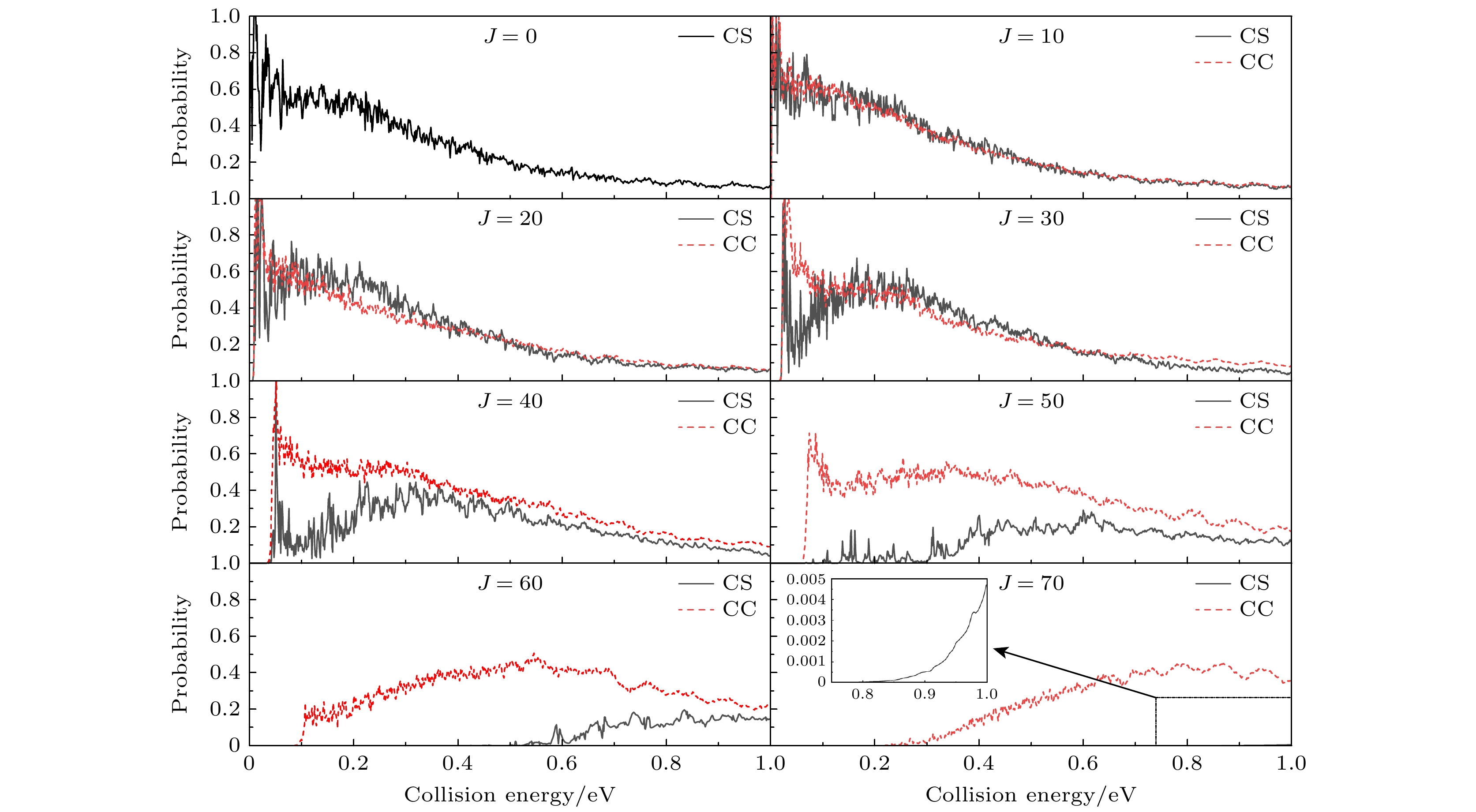
图 3
$ \text{H}({}^{2}\text{S})+\text{SiH}({\text{X}}^{2}\Pi )(\nu = 0, j = 0) $ 反应不同总角动量量子数(J = 0, 10, 20, 30, 40, 50, 60, 70)对应的反应概率随着能量的变化Figure 3. The reaction probabilities of CC and CS calculations for
$ \text{H}({}^{2}\text{S})+\text{SiH}({\text{X}}^{2}\Pi ) $ $ (\nu = 0, j = 0) $ reaction at J = 5, 10, 20, 30, 40, 50, 70.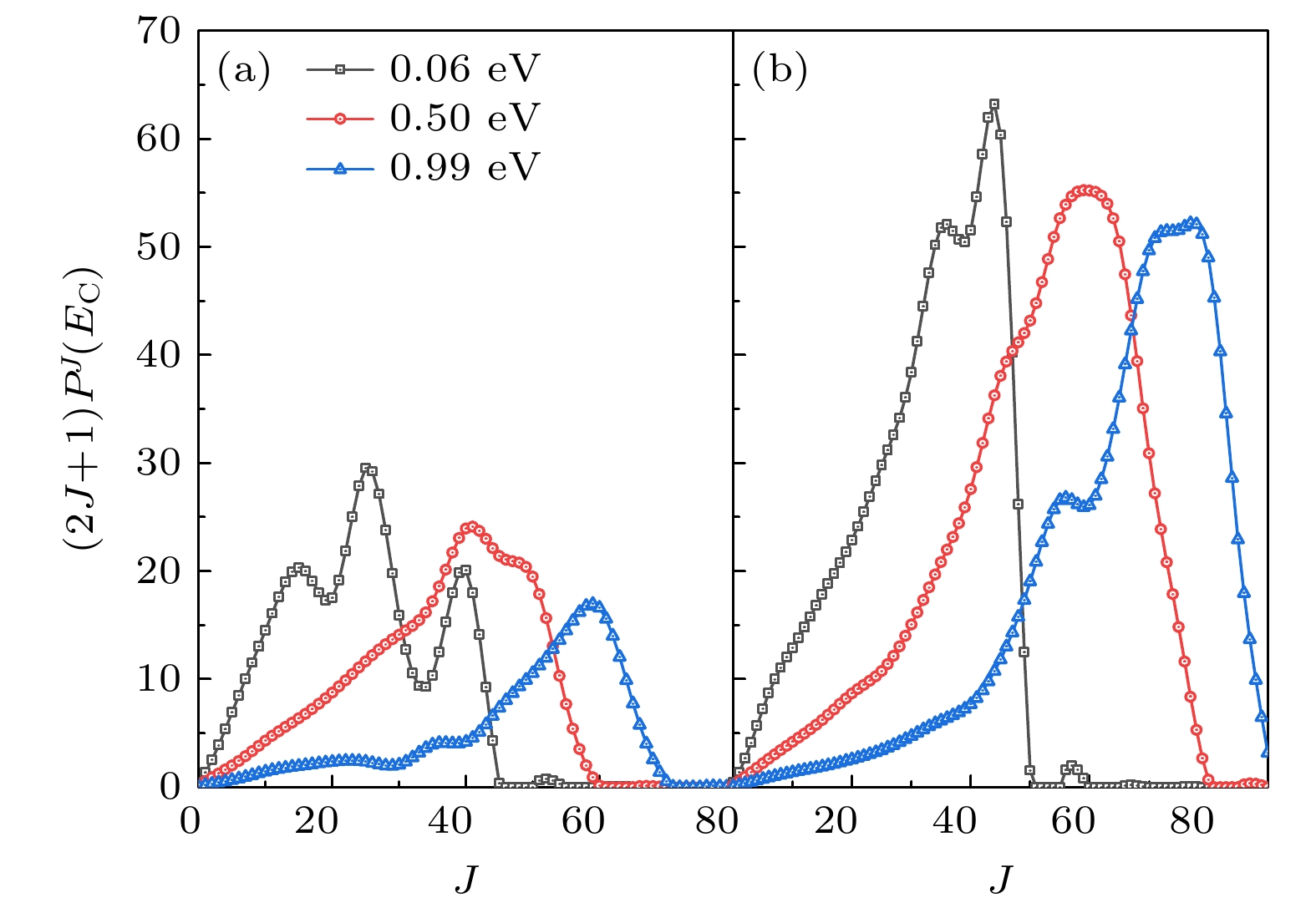
图 4 在不同能量Ec = 0.06, 0.50, 0.99 eV, 分波对于积分散射截面的贡献 (a) CS分波贡献; (b) CC分波贡献
Figure 4. Partial wave contributions to the integral cross section at Ec = 0.06, 0.50, 0.99 eV: (a) CS; (b) CC.
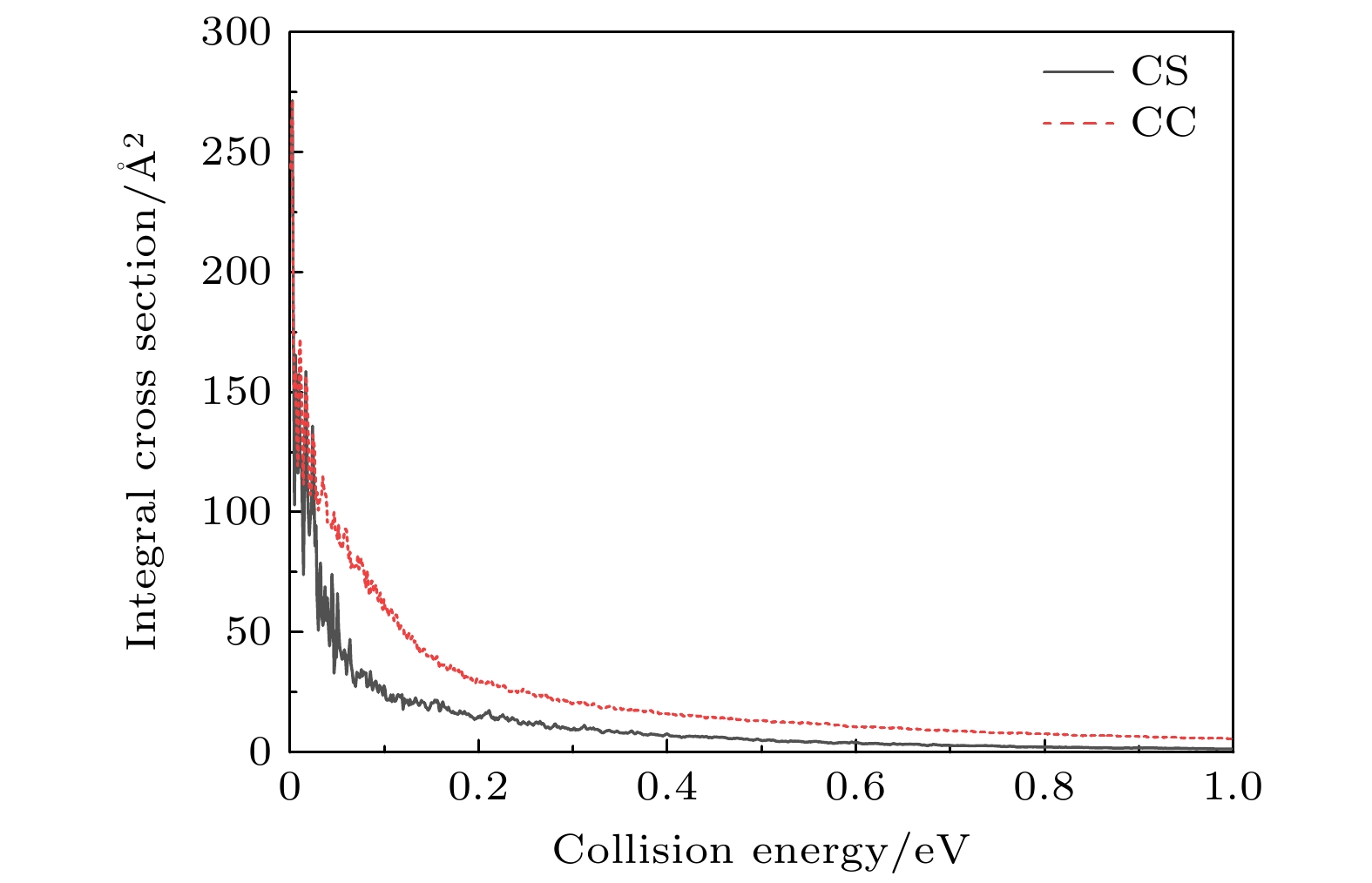
图 5 在
$ 1.0 \times {10^{ - 3}} $ —1.0 eV的碰撞能量范围下, H+ SiH反应ICS随着碰撞能量的变化Figure 5. The ICSs of CS and CC calculations for H+ SiH reaction versus collision energy of
$ 1.0 \times {10^{ - 3}} $ –1.0 eV.表 1 波包计算中的数值参量(采用原子单位a.u., 特殊情况另外注明)
Table 1. Model parameters of wave packet calculation (The atomic units are used in the calculation unless otherwise stated).
参量 H+SiH 散射坐标R的范围 (10–16, 22) 散射坐标R内格点数 383 内部坐标r的范围 (0.5, 16) 内部坐标r内格点数 255 角度γ范围 (90°, 180°) 角度格点数 200 阻尼起点Rd(rd) 18.0(14.0) 阻尼范围dR(dr) 0.0005(0.001) 初始波包的中心位置R0 16.0 初始波包的能量E0/eV 0.15 初始波包的宽度δ 0.3 光谱控制 0.1 流计算的位置rf 13.8 传播步数 100000  下载: 导出CSV
下载: 导出CSV
-
[1] Power D, Brint P, Spalding T 1984 J. Mol. Struct. 108 81 doi: 10.1016/0166-1280(84)80103-7 [2] Kalemos A, Dunning Jr T H, Mavridis A 2004 Mol. Phys. 102 2597 doi: 10.1080/00268970412331293802 [3] Ball J R, Thomson C 1978 Int. J. Quantum Chem 14 39 doi: 10.1002/qua.560140105 [4] Allen W D, Schaefer H F 1986 Chem. Phys. 108 243 doi: 10.1016/0301-0104(86)85046-7 [5] Jasinski J M, Chu J O 1988 J. Chem. Phys. 88 1678 doi: 10.1063/1.454146 [6] Herzberg G, Lagerqvist A, Mckenzie B J 1969 Can. J. Phys. 47 1889 doi: 10.1139/p69-240 [7] Dubois I, Herzberg G, Verma R D 1967 J. Chem. Phys. 47 4262 doi: 10.1063/1.1701609 [8] Dubois I 1968 Can. J. Phys. 46 2485 doi: 10.1139/p68-608 [9] Thoman Jr J, Steinfeld J 1986 Chem. Phys. Lett. 124 35 doi: 10.1016/0009-2614(86)85008-4 [10] Ishikawa H, Kajimoto O 1991 I. Mol. Spectrosc. 150 610 doi: 10.1016/0022-2852(91)90252-6 [11] Hirota E, Ishikawa H 1999 J. Chem. Phys. 110 4254 doi: 10.1063/1.478308 [12] Yurchenko S N, Bunker P R, Kraemer W P, Jensen P 2004 Can. J. Chem. 82 694 doi: 10.1139/v04-030 [13] Tokue I, Yamasaki K, Nanbu S 2005 J. Chem. Phys. 122 144307 doi: 10.1063/1.1876112 [14] Tokue I, Yamasaki K, Nanbu S 2006 J. Chem. Phys. 124 114308 doi: 10.1063/1.2183301 [15] Wu Y N, Zhang C F, Ma H T 2017 RSC Adv. 7 12074 doi: 10.1039/C7RA01021D [16] Cao J W, Wu Y N, Ma H T, Shen Z T, Bian W S 2021 Phys. Chem. Chem. Phys. 23 6141 doi: 10.1039/D0CP05540A [17] Wang H N, Lü Y L, Chen J X, Song Y Z, Zhang C Y, Li Y Q 2022 Phys. Chem. Chem. Phys. 24 7759 doi: 10.1039/D1CP05432E [18] Skouteris D, Castillo J F, Manolopoulos D E 2000 Comput. Phys. Commun. 133 128 doi: 10.1016/S0010-4655(00)00167-3 [19] Bulut N, Castillo J F, Jambrina P G, Kłos J, Roncero O, Aoiz F J, Bañares L 2015 J. Phys. Chem. A 119 11951 doi: 10.1021/acs.jpca.5b00815 [20] Chu T S, Zhang Y, Han K L 2006 Int. Rev. Phys. Chem. 25 201 doi: 10.1080/01442350600677929 [21] Lagana A, Lendvay G 2005 Theory of Chemical Reaction Dynamics (New York : Springer) p217 [22] Lin S Y, Guo H 2003 J. Chem. Phys. 119 11602 doi: 10.1063/1.1624060 [23] Lin S Y, Guo H 2004 J. Phys. Chem. A 108 2141 doi: 10.1021/jp031184h [24] Gao F, Wang X L, Zhao W L, Song Y Z, Meng Q T 2018 Eur. Phys. J. D 72 224 doi: 10.1140/epjd/e2018-90144-5 [25] Gao F, Zhang L L, Zhao W L, Meng Q T, Song Y Z 2019 J. Chem. Phys. 150 224304 doi: 10.1063/1.5088637 [26] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 103 2903 doi: 10.1063/1.470477 [27] Mandelshtam V A, Taylor H S 1995 J. Chem. Phys. 102 7390 doi: 10.1063/1.469051 [28] Tal-Ezer H, Kosloff R 1984 J. Chem. Phys. 81 3967 doi: 10.1063/1.448136 [29] Neuhauser D, Baer M, Judson R S, Kouri D J 1990 J. Chem. Phys. 93 312 doi: 10.1063/1.459603 [30] Althorpe S C 2001 J. Chem. Phys. 114 1601 doi: 10.1063/1.1334866 [31] Zhai H C, Lin S Y 2015 Chem. Phys. 455 57 doi: 10.1016/j.chemphys.2015.04.012 [32] Zhang L L, Liu D, Yue D G, Song Y Z, Meng Q T 2020 J. Phys. B: At. , Mol. Opt. Phys. 53 095202 doi: 10.1088/1361-6455/ab7641 [33] Harding L B, Guadagnini R, Schatz G C 1993 J. Phys. Chem. 97 5472 doi: 10.1021/j100123a005 [34] Peng Y, Jiang Z A, Chen J S 2017 J. Phys. Chem. A 121 2209 doi: 10.1021/acs.jpca.6b12125 [35] Peng Y, Zhang H 2022 J. Phys. Chem. A 126 1946 doi: 10.1021/acs.jpca.1c10860 [36] Buren B, Zhang J P, Li Y Q 2024 J. Phys. Chem. A 128 5115 doi: 10.1021/acs.jpca.4c01891 -
计量
- 文章访问数: 491
- HTML全文浏览数: 491
- PDF下载数: 4
- 施引文献: 0