
 首页
首页 登录
登录 注册
注册



-
为了面对全球气候变化, 目前超过150个国家做出碳中和承诺, 将近50%的碳中和技术转让发生在能源领域, 而储能是实现能源革命的关键支撑技术[1,2]. 因此, 急需创造更多的储能技术来匹配我们快速变化的世界. 近年来的研究与发现, 锂离子电池(LIBs)具有高能量密度、长周期寿命、轻巧便携、环境友好的优点, 使得其成为便携式设备和电动汽车中最合适的电能存储装置[3,4]. 然而, 由于全球能源绿色转型和锂资源的有限性, 使得近年来锂的价格急速上涨, 积极开发新型储能技术已成为必然趋势[5].
锂、钠和钾同属第一主族元素, 具有相似的化学性质. 这一特性促进了钠离子电池(SIBs)和钾离子电池(KIBs)的发展. 与锂离子电池相比, 钠离子电池和钾离子电池具有显著的资源和成本优势. 地壳中锂的平均含量(质量分数)仅为0.0017%, 且分布不均衡, 相比之下, 钠和钾的含量(质量分数)分别高达2.3%和 1.5%, 远高于锂[6]. 这使得钠离子电池和钾离子具有较好的成本优势, 更适合用于大规模储能系统. 尽管近年来许多研究报道钠离子电池和钾离子电池是锂离子电池潜在替代品, 然而高性能的负极材料的开发和实际需求却相差甚远[7,8].
目前, 钠/钾离子电池的研究主要集中在制备合适的正极材料上, 而负极材料的发展则要缓慢得多[9]. 作为电池中的关键组成部分, 负极材料的选择直接影响到电池的电化学性能和循环寿命. 但目前的钠/钾离子电池负极材料的相关研究主要集中在碳基材料和IVA, VA族金属化合物[9]. 如石墨作为商用锂离子电池最常见的负极材料, 具有长程有序的层状结构[10]. 石墨存储Li和K的可逆比容量分别为372 mAh/g和273 mAh/g, 然而存储Na的容量低至35 mAh/g[6], 限制了其在钠离子电池中的应用. 硬碳作为最主流的钠离子电池负极材料, 不同前驱体制备的硬碳具有不同的特性、形貌和结构, 这使得不同结构的硬碳材料对金属离子的存储能力存在较大差异, 且目前大部分的硬碳负极材料的比容量在300 mAh/g左右[11,12]. 如Kuai等[13]制备的生物质硬碳作为钠离子电池负极材料具有303.8 mAh/g的可逆比容量. Zhang等[14]通过龙虾壳制备的硬碳材料作为钾离子电池负极可逆比容量为277 mAh/g. 虽然金属复合负极材料表现出较高的电化学活性, 但往往伴随着较大的体积变化, 导致循环稳定性差[15–17]. 因此, 寻找高性能且稳定负极材料对于这些可充电电池的基本应用起着至关重要的作用.
相较于3D材料, 2D材料以其独特的2D结构和优异的电化学性能脱颖而出[18]. 例如, 在最近的研究发现, 二维磷烯作为钠和钾离子电池的负极理论比容量均高达865 mAh/g[19], 碳化物MXenes材料V2C, Cr2C, MoC2, TaC等碳化物MXenes材料作为锂离子电池负极材料的理论比容量分别为940, 1386, 894, 556 mAh/g[20–23]. 目前石墨烯及其衍生物、磷烯、过渡金属碳化物、氮化物和碳氮化物等二维材料在金属离子电池中均得到了广泛的探索[11,12,24–26]. 石墨烯作为使用最广泛的二维材料, 其具有柔韧性、高导电性、高比表面积[27,28], 例如, 石墨烯的储Li理论比容量高达744 mAh/g. 实际上本征石墨烯的储锂能力与其理论值相差甚远, 甚至逊色于石墨[29]. 因为Li原子在石墨烯层上容易形成团簇, 导致枝晶的形成, 从而使得循环过程中会使得容量衰减较快[30,31]. Pollak等[32]通过原位拉曼光谱研究发现, 锂与碳的结合能较低以及石墨烯吸附的Li原子之间存在着较强的库仑排斥作用, 很可能导致单层石墨烯的表面锂覆盖率较低.
幸运的是通过表面修饰可以增强石墨烯与Li/Na/K的相互作用, 掺杂剂可以有效地调节C原子的电荷和自旋密度, 从而调节石墨烯的电荷分布[33,34]. N和B作为C的临近元素, 它们有着接近的原子尺寸, 故N和B成为石墨烯最常见的改性掺杂剂. 目前大量研究表明N掺杂石墨烯是一种很有前途的负极材料[35–38]. Ma等[38]利用第一性原理计算了N掺杂石墨烯3种不同的模型在LIBs中的应用, 研究发现吡啶型石墨烯最适合储锂, 理论比容量高达1262 mAh/g. Cai等[39]通过实验合成N, S共掺杂石墨烯片作为LIBs负极材料在100 mA/g的电流密度下可逆容量达到1016 mAh/g. 值得注意的是, 单原子金属掺杂石墨烯近年成为催化领域研究的热点, 这是由于单空位掺杂增强石墨烯反应活性, 进而提升了催化反应效率[40,41]. Xiao等 [42]通过第一性原理计算发现石墨烯基单原子催化剂(SACs)一定程度上可以改变Li2S的绝缘性, 并降低Li2S沉积/溶解过程的能垒. 其中SA-Cu吸附Li2S后电子态密度在费米能级(EF)附近最高, 并且可有效的催化Li2S的氧化还原反应. 但目前将单原子金属掺杂石墨烯作为电极材料的研究相对有限.
为了探究掺杂石墨烯的储能的更多可能性, 选择将吡啶(pyridinic)结构的石墨烯中的部分N替换为O, 并掺杂Cu得到Cu/NO2G材料. 本文采用第一性原理研究的方法确定Cu/NO2G在热力学和动力学上是稳定的, 并在吸附Li, Na, K前后均保持良好导电性. 随后, 通过增加Cu/NO2G上吸附Li/Na/K原子的浓度来确定材料的储存能力, 结果显示Cu/NO2G具有成为高容量离子电池负极材料的潜力, 其储存Li, Na, K的理论比容量分别高达1639.9 mAh/g, 2025.8 mAh/g, 1157.60 mAh/g. 且在Li, Na, K离子嵌入过程中, Cu/NO2G的晶格常数变化均小于1%, 这意味着其具有良好的循环稳定性. 最后, 评估了Li/Na/K在Cu/NO2G表面的扩散行为, Li, Na, K离子在Cu/NO2G表面的迁移势垒分别为0.339 eV, 0.209 eV和0.098 eV, 表明其优异的倍率性能. 以上结果都表明Cu/NO2G是一种高性能的碱金属离子电池负极.
-
本文的所有计算都是在维也纳从头算模拟包(VASP)[43,44]软件下基于密度泛函理论(DFT)[45,46]完成的. 本文的交换关联势采用局域密度近似(LDA)用Perdew和Zunger参数化的Ceperly-Alder泛函来模拟[47,48]. 为了确保计算的准确性, 平面波展开截止能量Ecut设置为520 eV, 采用Monkorst-Pack方案对布里渊区进行采样[49], 结构优化布里渊区K点采样网格数为3 × 3 × 1, 电子结构计算布里渊区K点采样网格数为5 × 5 × 1. 总能量和离子力的收敛标准分别为10–5 eV和10–3 eV/Å. 声子谱使用phonopy 软件包进行计算[50]. 确定碱性金属原子在Cu/NO2G的最佳迁移路线和势垒, 具体是采用CI-NEB方法[51]在包含4个Cu原子、4个N原子、8个O原子和56个C原子的2 × 2超胞上进行计算. 在所有计算中, 沿材料平面法向量方向的真空层都确保高于20 Å, 以避免每个片层的相互作用.
-
首先, 用1个N原子和2个O原子取代由17个C原子组成的缺陷石墨烯空位处的3个C原子, 并在缺陷空位掺杂1个Cu原子, 掺杂Cu原子的屈曲高度为1.412 Å. 图1(a), (b)为Cu/NO2G结构优化后的俯视图和侧视图. 该结构的晶格常数a = b = 7.332 Å , Cu—N, Cu—O, N—C键长分别为1.886 Å, 2.070 Å和1.344 Å, C—C平均键长为1.412 Å, O—C平均键长为1.390 Å. 由于材料中掺杂了Cu原子, 材料的两面不再具有对称性, 因此如图1(b)中, 将材料凸面标记为A面, 凹面标记为B面. 此外, 如图1(c), (d)所示, 色散声子谱和分子动力学计算也表明材料在热力学和动力学上具有稳定性, 这为Cu/NO2G作为离子电池电极材料提供了保障.
-
离子电极材料的一个关键特性是对碱金属离子具有较强的吸附能力. 为了确定Li, Na和K在Cu/NO2G 的最佳吸附位置, 本研究系统地考察了图2(a)所示的3种类型的吸附位点: 绿色为六圆环的中心上方的空位、青色为原子上方的顶位、粉色为原子与原子之间上方的桥位. 鉴于材料两面的不对称性, 吸附计算需要分别考虑A面和B面. 吸附能Ead的计算公式定义如下:
其中Ehost, Etotal分别为单个A (A = Li/Na/K)原子吸附在Cu/NO2G前后的系统总能,
$ {E}_{A} $ 为碱性金属A的内聚能. 根据(1)式, 吸附能应为负值, 这有助于抑制枝晶的形成. 吸附能越小, 表明相应的吸附结构越稳定, 且有利于克服金属离子之间的库仑排斥作用, 从而实现电池的高容量.Li和Na在Cu/NO2G的A面最稳定的吸附点为C5O环的空位的上方, K 在Cu/NO2G的A面最稳定的吸附点为C6环的空位的上方, 吸附后构型如 图2(b)—(d), 吸附能分别为–0.508 eV , –0.442 eV和–0.940 eV. Li (0.76 Å)和Na (1.02 Å)的离子半径较小, 更容易适应孔径较小的C5O (C—C平均键长为1.395 Å; C—O平均键长为1.399 Å)环, 与其形成更紧密的相互作用. 相较之下, K (1.38 Å)的离子半径较大, 更适合孔径更大的C6环(C—C平均键长为1.423 Å). 如图2(e)—(g)所示, B面Li, Na和K最稳定的吸附点则均为Cu原子的顶位附近, 吸附能分别为–1.434 eV, –0.844 eV和–1.197 eV. Bader电荷分析显示Cu/NO2G吸附碱金属原子后, Li, Na和K均与石墨烯层之间存在电荷转移, Li向Cu/NO2G单层转移了0.845e, Na向Cu/NO2G单层转移了0.835e, K向Cu/NO2G单层转移了0.842e. 表1中的数据通过Bader电荷分析得到的不同元素的平均电子转移数量, 可以发现无论吸附哪种碱金属原子, Cu和C均失去电子带正电荷, 而N和O则是得到电子, 表现出电负性. 图3中的差分电荷密度图直观地展示了吸附过程中的电荷得失情况.
为了确认材料是否在吸附碱性金属前后均保持导电性, 因此选择计算了本征Cu/NO2G吸附Li/Na/K后最稳定的吸附构型的态密度(DOS), 如图4(a)所示, 可以看到本征Cu/NO2G呈现金属态, 费米能级附近的态密度主要由C贡献. 考虑到Cu/NO2G中的碳(C)原子数量远大于其他原子(如Cu, O等), 碳的态密度会在总体态密度中占有较大的比例. 随后绘制了局部态密度(LDOS)图, 如图4(b), (c)所示, C的2p轨道态密度在费米能级附近最为突出, Cu的4s轨道在费米能级附近也较为突出, 说明在Cu的掺杂对提高材料的导电性有一定的贡献. 如图4(d)—(f)所示, 当吸附Li/Na/K时, 材料仍然保持着金属态, 这可以说明Cu/NO2G若是作为LIBs, SIBs和KIBs的负极材料, 可以保证电池在运行过程中具有良好的导电性.
-
本研究进一步计算了Cu/NO2G嵌入Li/Na/K的平均开路电压 (OCV)和理论比容量, 对于可充电离子电池的负极材料, 高比容量和适宜的工作电压是其关键性能指标. 平均OCV是阳极的理论氧化还原电位, 从安全和能量密度的角度看, 较低的平均OCV值对阳极材料更为有利. 根据3.2节单个Li/Na/K原子吸附的基础上在Cu/NO2G上增加Li/Na/K原子的吸附浓度, 相应的半电池反应方程式如下:
其中, A为碱性金属原子Li/Na/K, z为Cu/NO2G上吸附的Li/Na/K原子的数量. 为了评估吸附原子与宿主材料之间的相互作用的大小, 定义平均吸附能Eave的计算公式如下:
其中Ehost, EA的定义与上文保持一致, Etotal为Cu/NO2G吸附多个Li/Na/K原子后的体系总能量. Eave为负值时, 表明碱性金属原子具有自发嵌入的可能性. 因此当Eave > 0时, 即可推断材料对碱金属原子的吸附能力已达到其饱和状态, 即金属原子吸附量已达峰值. 忽略体积和熵效应, 平均开路电压Vocv采用如下公式[52,53]进行计算:
考虑到实际离子在充放电过程中迁移速度会非常快, 根据3.2节计算的吸附位点的稳定性前期按3个的增量增加Li, Na和K原子的浓度. 当 Cu/NO2G吸附3, 6, 9, 12, 15, 17个Li原子时Eave均为负值, 分别为–0.145 V, –0.217 V, –0.273 V, –0.078 V, –0.070 V, –0.039 V, 当吸附Li原子数增加到18个时, Cu/NO2G的Eave为正值(0.017 V). 因此Cu/NO2G最多可以吸附17个Li原子, 其对应的化学计量数为Li17CuNO2C14. Vocv与Li原子数量的折线图如图5所示. 对于在 Cu/NO2G上吸附的Na原子数量z = 3, 6, 9, 12, 15, 18, 21和24时, 对应的Eave分别为–0.361 V, –0.224 V, –0.131 V, –0.073 V, –0.105 V, –0.036 V, –0.102 V和–0.018 V. 虽然当吸附Na原子数量为24时Eave为负值, 但弛豫后结构不稳定, 晶格变化达到了15%, 因此预测一个Cu/NO2G晶胞最多可以容纳21个Na原子, 对应的化学计量数为Na21CuNO2C14. 当Cu/NO2G上吸附的K原子数量z = 3, 6, 9, 12和15时, Eave分别为–0.576 V, –0.491 V, –0.205 V, –0.270 V 和–0.076 V. 同样地当Cu/NO2G上吸附的K原子数量超过12个后材料晶格常数的变化超过了10%, 故Cu/NO2G最多可容纳的K原子数量为12个, 对应的化学计量数为K12CuNO2C14.
随后计算了理论比容量, 并与其他材料的理论比容量进行了对比. 理论比容量计算公式如下:
其中z为吸附Li/Na/K原子的个数, F为法拉第常数 (26789 mAh/mol), M为Cu/NO2G的摩尔质量(
$ {M}_{{\mathrm{C}}{\mathrm{u}}/{\mathrm{N}}{{\mathrm{O}}}_{2}{\mathrm{G}}} $ =277.7 mol/g). 计算得到Cu/NO2G的储Li的理论比容量为1639.9 mAh/g, 对应的晶格变化约为0.61%. 其理论比容量均高于吡啶 (pyridinic)结构的石墨烯的理论比容量, 更是石墨 (约372 mAh/g)[54]储Li容量的4倍多. 而Cu/NO2G储Na的理论比容量更是高达2025.8 mAh/g, 晶格变化也只有~0.19%. Cu/NO2G 储K的理论比容量为1157.6 mAh/g, 晶格变化为约0.92%. 可见Cu/NO2G作为锂/钠/钾离子电池的负极材料不仅拥有优秀的理论比容量, 还具备出色的循环性能.如图6所示, 将Cu/NO2G的理论比容量与近年来发现二维材料的理论比容量进行对比, 可以发现Li, Na和K的理论比容量仅仅低于b-Bi而高于其他二维材料[52]. 其中Cu/NO2G储Na理论比容量高于B2S (~1498 mAh/g)[55,56], PC6 (~1301 mAh/g)[57], BP (~1282 mAh/g)[58,59], Si3C (~1115 mAh/g)[60] , 是Ti2CS2 (~936 mAh/g)[61], Phosphorene (~865 mAh/g)[62,63], MoN2 (~864 mAh/g) [64], TiSi2N4 (~854 mAh/g)[65], GeSe (~707 mAh/g)[66], DB silicene(~622 mAh/g)[67], V3C2(~606 mAh/g)[68], TiB4 (~588 mAh/g)[69], GeS (~512 mAh/g)[70]的理论比容量的2—3倍, 甚至是TiS2 (~479 mAh/g)[71], Ti3C2S2 (~463 mAh/g)[72], Ti3C2 (~352 mAh/g)[73], Nb2S2C (~348 mAh/g)[74], SrB8 (~308 mAh/g)[69], Nb2C (~271 mAh/g)[75], VS2 (~233 mAh/g)[76,77]的理论比容量的4倍以上.
特别注意的是, Cu/NO2G的储锂、储钠和储钾的理论比容量都比较高, 这主要是因为Cu/NO2G体系中, 掺杂原子Cu可以调控石墨烯的电子结构和表面性质, 此外锚定原子, 即氮原子和氧原子可以使得活性位点所在位置的面积增大, 形成协同吸附效应进而进一步增加活性位点的活性, 进一步提升对碱金属离子的吸附能力从而获得提升存储容量. 另外, 储锂、储钠和储钾的理论比容量具有差异, 这种差异性的可能性主要归因于以下两点. 1) 离子吸附能力: 不同碱金属离子的电负性不同, 与基底材料的相互作用强度也不同, 导致吸附能存在差异. 吸附能越负, 吸附能力越强, 理论比容量越高. 钠离子在更多吸附位点上具有负的吸附能, 且吸附能普遍低于锂和钾, 表明其与基底材料的相互作用更强, 因此具有更高的理论比容量. 2) 离子尺寸和结构稳定性: 钾的离子半径最大, 其次是钠, 锂最小. 较大的离子尺寸会导致更大的晶格常数的变化, 并且在吸附数量较多时有更强的静电排斥效应, 从而影响材料的结构稳定性导致存储能力减弱. 例如尽管钾的吸附能力强于锂, 但我们的计算表明, 锂的理论比容量高于钾的, 这主要是因为当吸附钾的数量达到13时, 尽管平均吸附能仍为负值, 但材料的体积膨胀过大, 显著削弱了结构的稳定性. 总之, Cu/NO2G的储锂、储钠和储钾的存储能力的差异受到材料结构、离子性质等多种因素的影响.
-
扩散势垒与电池的倍率性能密切相关, Li/Na/K离子在材料上的扩散势垒越低, 阳离子迁 移越快. 这里采用了CL-NEB[51]方法分别去求得Li/Na/K原子在Cu/NO2G上的鞍点、最低扩散势垒的路径. 基于Li/Na/K原子在Cu/NO2G单层的吸附最佳吸附点来确定迁移路径, 但由于两种材料的A和B侧的不对称性, 因此材料的A和B两侧均需要进行扩散计算, 这里采用2 × 2的超胞计算迁移势垒, 相应的化学计量数为Cu4N4O8C56.
首先计算Li/Na/K原子在Cu/NO2G单层A侧的迁移, Li/Na/K原子均考虑了3种迁移路线, 由于前面计算表明Li和Na单原子在Cu/NO2G的A侧最佳吸附位点为图2(a)的A1点, 而K原子在Cu/NO2G的A侧最佳吸附位点为图2(a)的A2点, 故K选择的迁移路径与Li/Na的迁移路径不同. 如图7(c)所示, Li原子在Path-1, Path-2和Path-3上的扩散势垒分别为0.339 eV, 0.649 eV和1.254 eV, Na原子在Path-1, Path-2和Path-3上的扩散势垒分别为0.224 eV, 0.209 eV和0.826 eV, K原子在Path-1, Path-2和Path-3上的扩散势垒分别为0.098 eV, 0.297 eV和0.381 eV. 对于Cu/NO2G的B侧, 如图7(b)所示, Li/Na/K选择迁移路径相同, 对于Path-1和Path-2两种迁移路径, Li原子扩散势垒分别为1.959 eV 和1.758 eV, Na原子的扩散势垒分别为0.813 eV 和0.737 eV, K原子的扩散势垒分别为0.838 eV和 0.459 eV. 总之, 在Cu/NO2G单层的A侧, K原子的迁移最容易, 特别是在Path-1上, 扩散势垒仅为0.098 eV, 而Li原子的扩散势垒较高, 特别Path-3达1.254 eV. 在B侧, Li的迁移势垒最高(Path-1为1.959 eV), Na和K的势垒较低, 其中K在Path-2上的势垒最低, 仅为0.459 eV. 总体来看, 金属离子在Cu/NO2G不同侧面和迁移路径上的扩散势垒存在显著差异. 而Li和Na原子均是在Cu/NO2G的A侧迁移势垒最低, Li和Na在A侧的最低迁移势垒分别为0.339 eV和0.224 eV, K原子在A侧的Path-1的迁移势垒仅为0.098 eV. 而相较A侧, B侧的迁移势垒总体较高, 但K原子在Cu/NO2G的B侧的迁移势垒也低于Na和Li, 在B侧的Path-2的迁移势垒为0.459 eV, 这展示出Cu/NO2G作为KIBs电池负极材料, 具有较高性能的倍率性能. 这里可以发现, 随着碱性金属原子序数的增加, 扩散势垒呈现降低趋势, 这与Li < Na < K的原子半径增大基本一致. 碱性金属的这种行为与之前的报道一致[52,55–61].
初始空段落
-
综上所述, 本研究基于第一性原理计算探讨了N, O双掺杂石墨烯负载Cu原子的二维材料 (Cu/NO2G)作为锂离子电池、钠离子电池和钾离子电池负极材料的应用潜力. 研究结果揭示, Cu/NO2G满足良好的电极材料的必要要求: 1) Cu/NO2G吸附Li/Na/K前后均保持金属性; 2) Cu/NO2G展现出迄今所研究的绝大部分2D负极材料的优异金属离子存储能力, Li/Na/K的理论比容量分别达到1639.9 mAh/g, 2025.8 mAh/g, 1157.6 mAh/g; 3) 更为关键的是, 在Li/Na/K原子嵌入过程中晶格变化很小 (<1%)变形, 这有利于保持良好的循环稳定性能; 4) 此外, Li原子在Cu/NO2G单层扩散势垒仅为0.339 eV, Na原子为0.209 eV , 而K原子的扩散势垒更是低至0.098 eV, 确保了Cu/NO2G作为负极材料的倍率性能. 总体而言, Cu/NO2G显示出作为LIBs, SIBs和KIBs负极材料的巨大潜力, 这为未来的理论研究和实验开发提供了坚实的基础.
-
支撑本研究成果的数据集可在科学数据银行
https://www.doi.org/10.57760/sciencedb.j00213.00063 中访问获取.
氮氧锚定的单原子铜掺杂石墨烯作为碱离子电池负极的理论预测研究
Theoretical prediction of nitrogen-oxygen-anchored monatomic copper-doped graphene as an anode for alkaline ion batteries
-
摘要: 合理设计高容量的新型电极材料是进一步提高离子电池能量密度的关键. 石墨烯曾被认为是离子电池负极材料最有前景的候选者之一, 然而因纯的石墨烯与相应离子的相互作用较弱, 导致其理论比容量都不高. 基于此, 本文通过第一性原理评估氮氧(N, O)锚定的单原子铜掺杂石墨烯的二维材料Cu/NO2G作为锂/钠/钾离子电池负极的可行性. 计算结果显示, Cu/NO2G在热力学和动力学上都是稳定的, 在吸附Li/Na/K前后均保持良好导电性, 并且Cu/NO2G储存Li/Na/K的理论比容量分别高达1639.9 mAh/g, 2025.8 mAh/g, 1157.6 mAh/g, 在Li/Na/K嵌入的过程中, 其晶格常数变化微小(<1%), 这预示着其循环稳定性能佳. 此外, Li, Na, K在Cu/NO2G表面上的迁移势垒分别为0.339 eV, 0.209 eV和0.098 eV, 表明其具有优异的倍率性能. 综上所述, 本文结果为合理设计金属单原子掺杂石墨烯作为碱金属离子电池的新型负极材料奠定了坚实的理论基础. 本文数据集可在
https://doi.org/10.57760/sciencedb.j00213.00063 中访问获取.Abstract: Reasonably designing high-capacity novel electrode materials is key to further enhancing the energy density of ion batteries. Graphene has been considered one of the most promising candidates for anodes in ion batteries. However, the weak interaction between pure graphene and the corresponding ions results in a low theoretical capacity. Based on this, in this work the first-principles calculation is used to assess the viability of two-dimensional Cu/NO2G, a single-atom copper-doped graphene anchored by nitrogen and oxygen, as an anode material for Li/Na/K-ion batteries. The results show that Cu/NO2G is stable in terms of thermodynamics and kinetics. It maintains good conductivity before and after the adsorption of Li/Na/K, with theoretical capacities of 1639.9 mAh/g for lithium, 2025.8 mAh/g for sodium, and 1157.6 mAh/g for potassium. In the embedding process of Li/Na/K, the lattice constant changes minimally (less than 1%), indicating excellent cycling stability. Additionally, the migration energy barriers for Li, Na, and K on the surface of Cu/NO2G are 0.339 eV, 0.209 eV, and 0.098 eV, respectively, demonstrating its superior rate performance. In summary, these results provide a solid theoretical foundation for rationally designing metal single-atom doped graphene as a novel anode material for alkali metal ion batteries. All the data presented in this paper are openly available athttps://doi.org/10.57760/sciencedb.j00213.00063 .-
Key words:
- first-principles /
- ion batteries /
- graphene /
- doping .
-

-
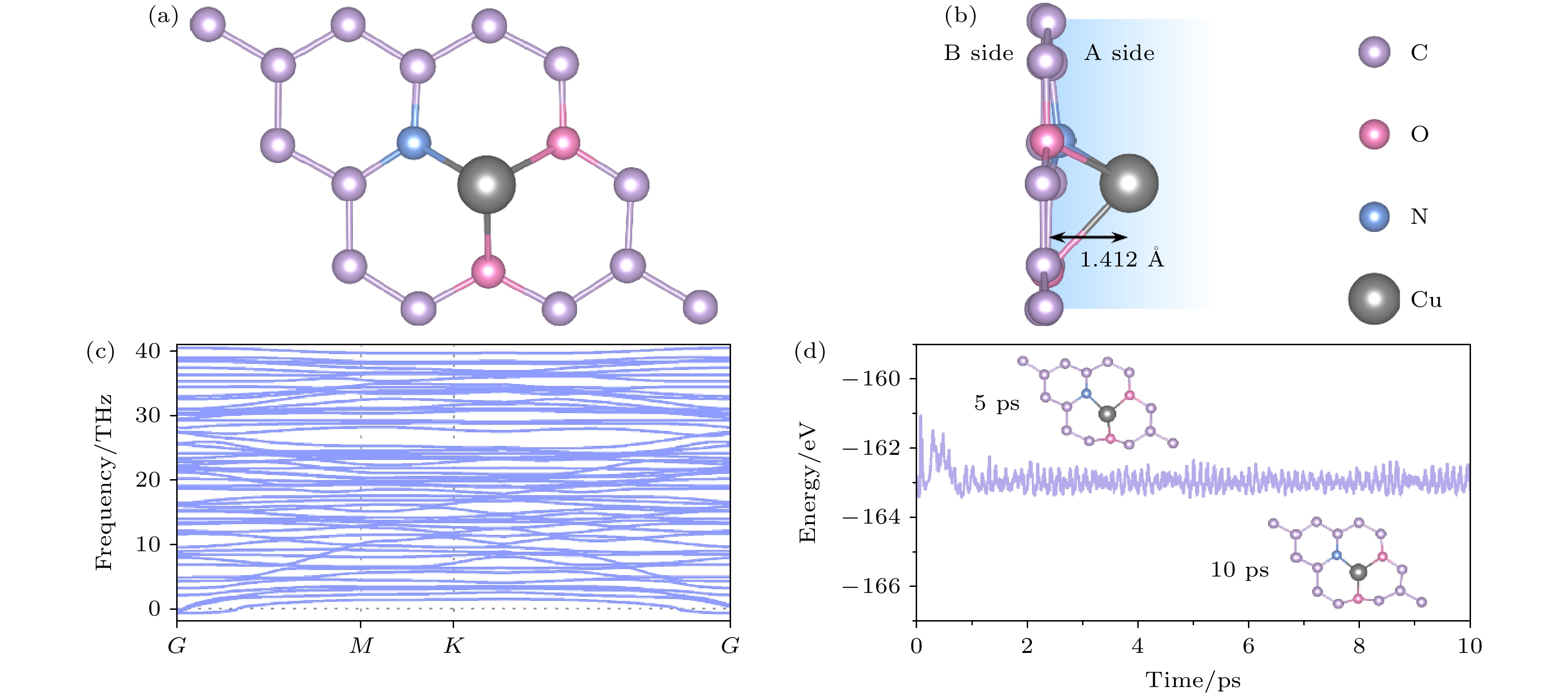
图 1 (a) 结构优化后Cu/NO2G的俯视图; (b) 结构优化后Cu/NO2G的侧视图; (c) Cu/NO2G单层的声子色散曲线; (d) 300 K下AIMD模拟的结果, 插图为5 ps和10 ps时的原子结构俯视图
Figure 1. (a) Top view of the optimized Cu/NO2G; (b) side view of the optimized Cu/NO2G; (c) phonon dispersion curves of Cu/NO2G monolayer; (d) results of AIMD simulations at 300 K, where insets are snapshots of atomic structures at 5 ps and 10 ps.
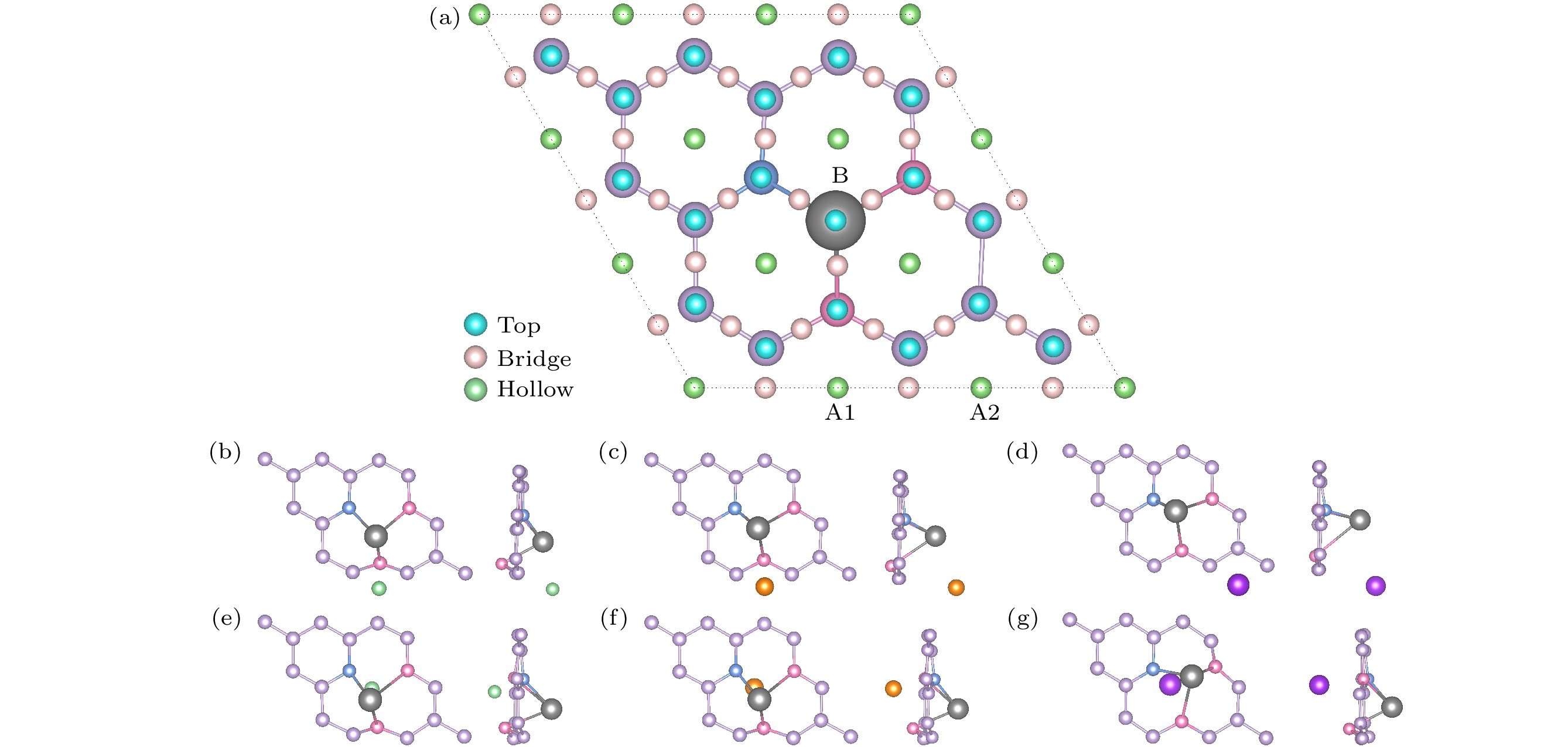
图 2 (a) Cu/NO2G上A面和B面所考虑的吸附位点俯视图; (b)—(d)分别为Li/Na/K在A面最稳定的吸附构型的俯视图和侧视图; (e)—(g) 分别为Li/Na/K在B面最稳定的吸附构型的俯视图和侧视图
Figure 2. (a) Top view of adsorption sites considered on side A and side B of Cu/NO2G; (b)–(d) the top and side views of the most stable adsorption configurations of Li/Na/K on the A side; (e)–(g) the top and side views of the most stable adsorption configurations of Li/Na/K on the B side.
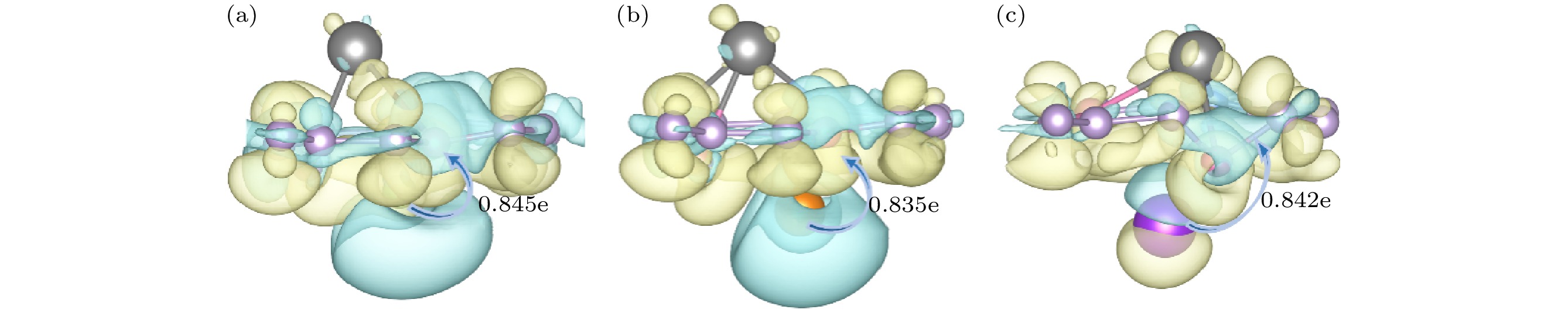
图 3 (a) Li, (b) Na, (c) K吸附在Cu/NO2G的B面上的电荷密度差的侧视图, 蓝色和黄色分别代表电子耗尽区和积累区, 相应的等值面(isosurface level)的数值为0.0015 Bohr–3
Figure 3. Side views of the charge density difference of (a) Li, (b) Na and (c) K atom adsorbed on B side of Cu/NO2G. The blue and the yellow colors represent regions with electron depletion and accumulation, respectively. The corresponding value of isosurface level is 0.0015 Bohr–3.
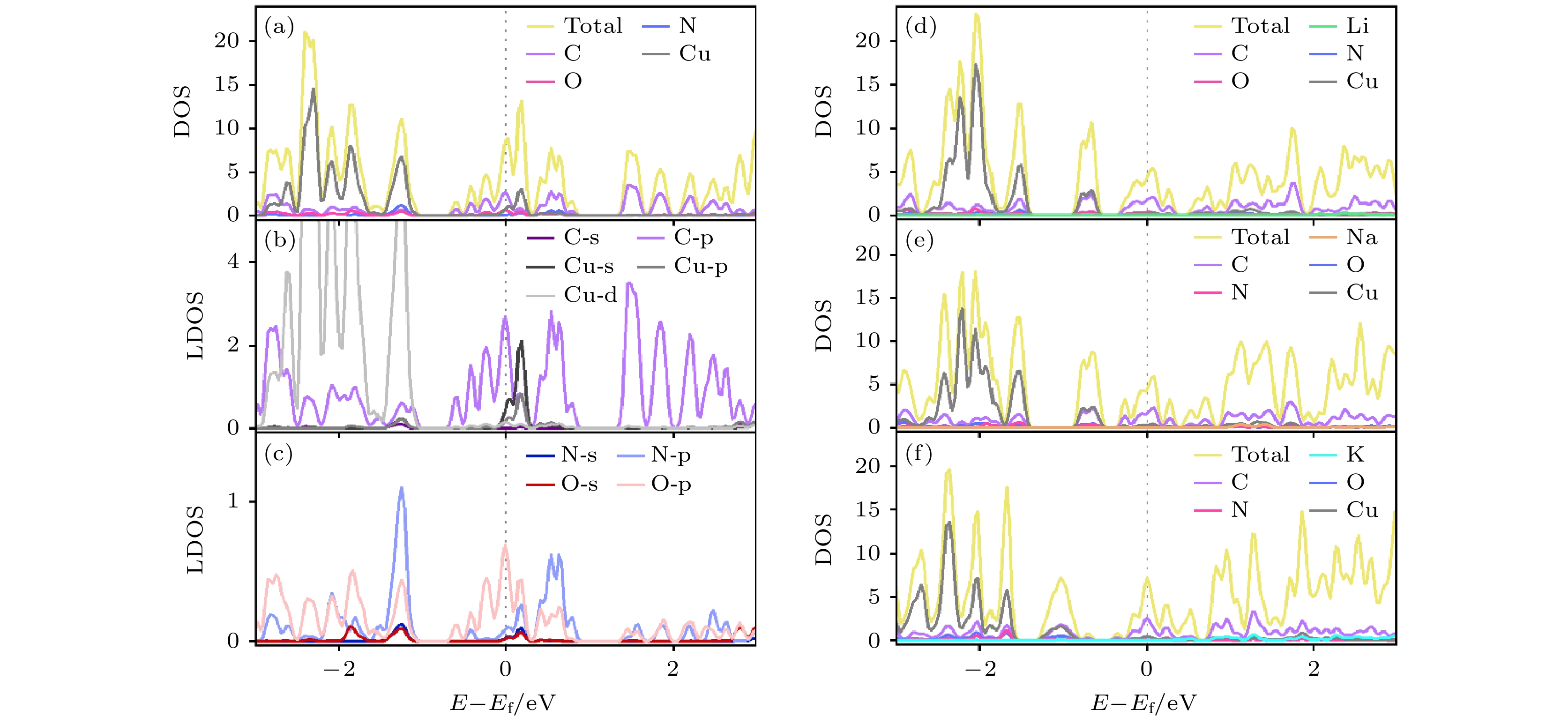
图 4 (a) Cu/NO2G态密度图; (b) Cu/NO2G 中C和Cu的局部态密度图; (c) Cu/NO2G 中N和O的局部态密度图; (d) Cu/NO2G吸附Li原子后态密度图; (e) Cu/NO2G吸附Na原子后态密度图; (f) Cu/NO2G吸附K原子后态密度图
Figure 4. (a) DOS of Cu/NO2G; (b) LDOS plots for C and Cu in Cu/NO2G; (c) LDOS plots for N and O in Cu/NO2G; (d) DOS of Cu/NO2G after Li adsorption; (e) DOS of Cu/NO2G after Na adsorption; (f) DOS of Cu/NO2G after K adsorption.
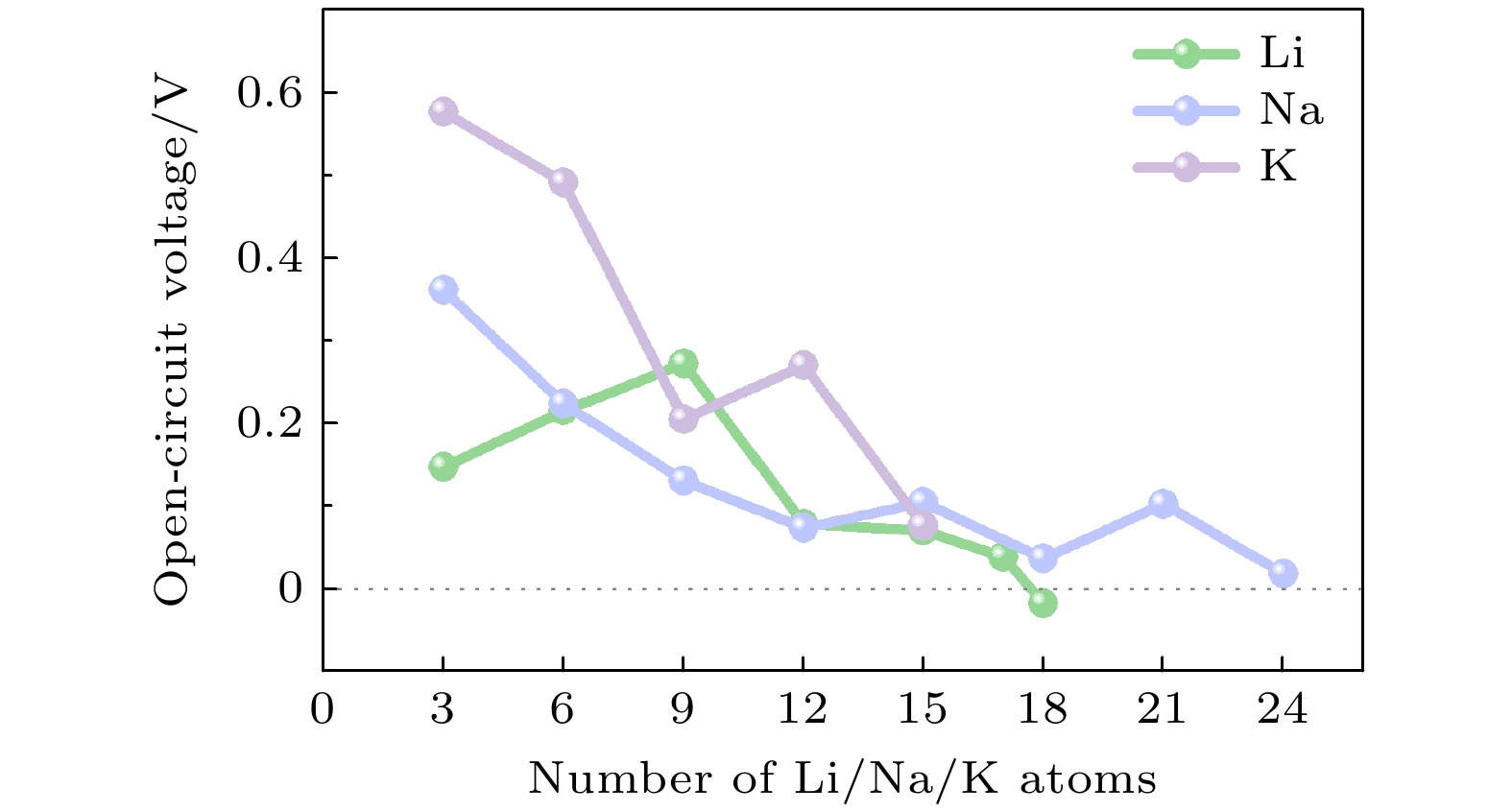
图 5 不同浓度Li/Na/K嵌入Cu/NO2G的
$ {V}_{{\mathrm{o}}{\mathrm{c}}{\mathrm{v}}} $ Figure 5. Vocv of the Cu/NO2G with different concentrations of Li/Na/K embedded.
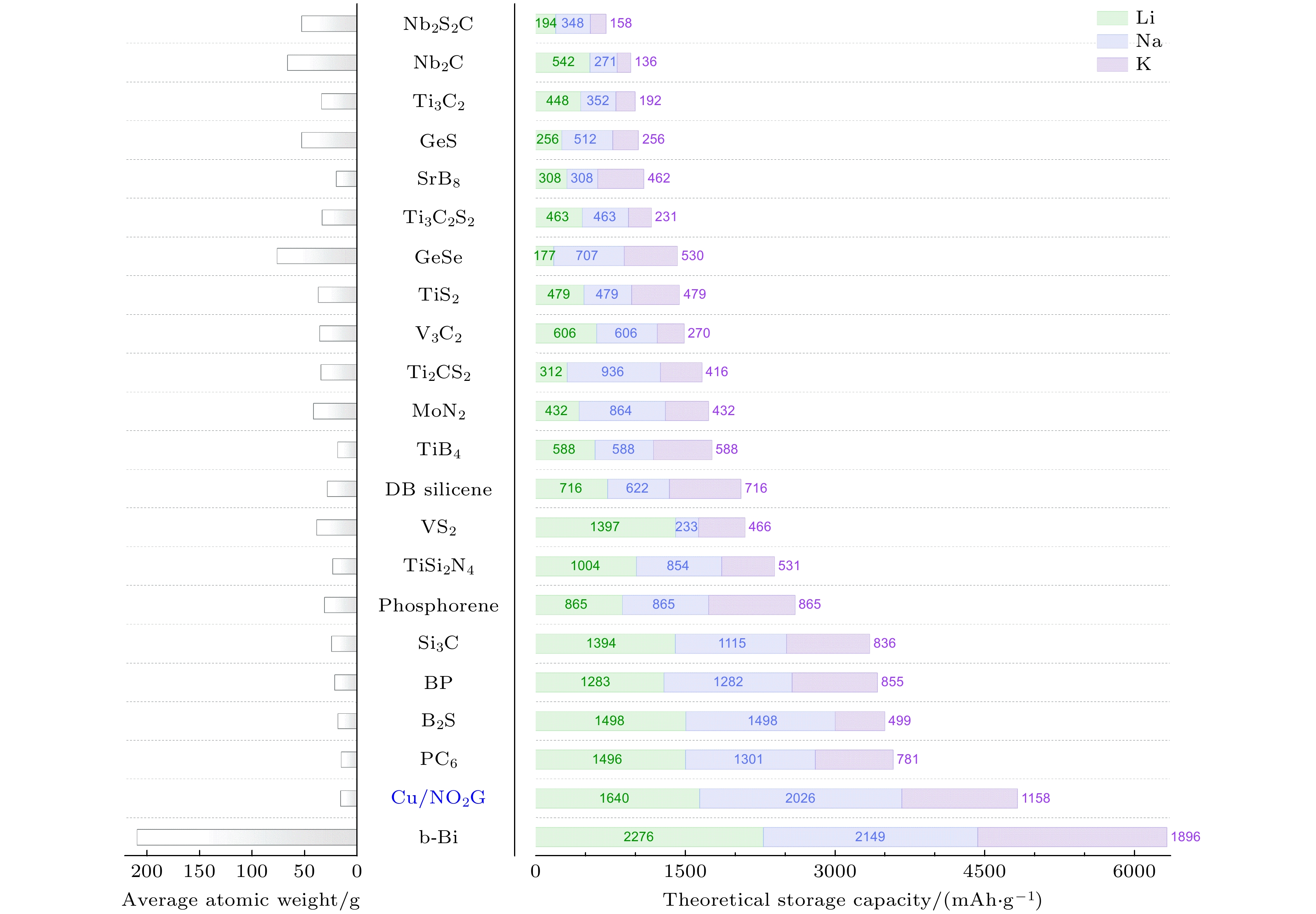
图 6 Cu/NO2G与其他二维材料的理论比容量对比图
Figure 6. Comparison of the theoretical specific capacities between Cu/NO2G monolayer and other 2D anode materials.
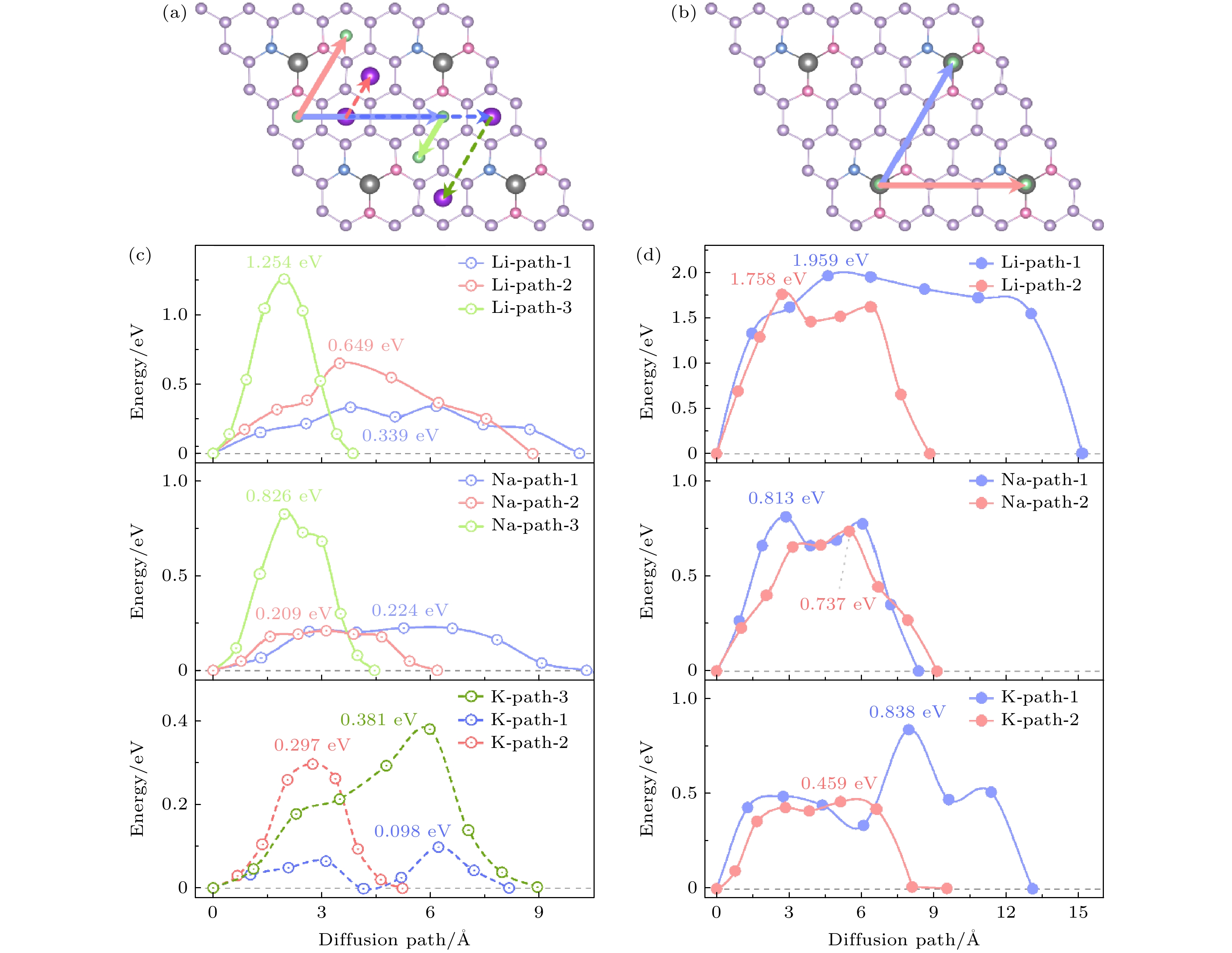
图 7 (a) Li/Na/K在Cu/NO2G的A面迁移路径; (b) Li/Na/K在Cu/NO2G的B面迁移路径; (c) Li/Na/K在Cu/NO2G的A面迁移路径上的扩散势垒; (d) Li/Na/K在Cu/NO2G的B面迁移路径上的扩散势垒
Figure 7. (a) Migration pathways of Li/Na/K on side A of Cu/NO2G; (b) migration pathways of Li/Na/K on side B of Cu/NO2G; (c) diffusion barriers of Li/Na/K for the pathways on side A of Cu/NO2G; (d) diffusion barriers of Li/Na/K for the pathways on side B of Cu/NO2G.
表 1 Cu/NO2G 吸附Li/Na/K后的Bader电荷分析
Table 1. Bader charge analysis of Cu/NO2G Li/Na/K adsorbed states.
Average charge states Li Na K C N O Cu $ {{\mathrm{L}}{\mathrm{i}}{\mathrm{C}}{\mathrm{u}}{\mathrm{N}}{{\mathrm{O}}}_{2}{\mathrm{C}}}_{14} $ –0.845 — — –0.162 1.232 1.115 –0.355 $ {{\mathrm{N}}{\mathrm{a}}{\mathrm{C}}{\mathrm{u}}{\mathrm{N}}{{\mathrm{O}}}_{2}{\mathrm{C}}}_{14} $ — –0.835 — –0.154 1.206 1.097 –0.403 $ {{\mathrm{K}}{\mathrm{C}}{\mathrm{u}}{\mathrm{N}}{{\mathrm{O}}}_{2}{\mathrm{C}}}_{14} $ — — –0.842 –0.142 1.224 1.053 –0.503  下载: 导出CSV
下载: 导出CSV
-
[1] Liu A, Chen Y T, Cheng X 2022 Environ. Res. Lett. 17 054031 doi: 10.1088/1748-9326/ac6819 [2] 陈海生, 刘畅, 徐玉杰, 岳芬, 刘为, 俞振华 2021 储能科学与技术 10 1477 doi: 10.19799/j.cnki.2095-4239.2021.0389 Chen H S, Liu C, Xu Y J, Yue F, Liu W, Yu Z H 2021 Energy Storage Sci. Technol. 10 1477 doi: 10.19799/j.cnki.2095-4239.2021.0389 [3] Nzereogu P U, Omah A D, Ezema F I, Iwuoha E I, Nwanya A C 2022 Appl. Surf. Sci. Adv. 9 100233 doi: 10.1016/j.apsadv.2022.100233 [4] Hossain M H, Chowdhury M A, Hossain N, Islam M A, Mobarak M H 2023 Chem. Eng. J. Adv. 16 100569 doi: 10.1016/j.ceja.2023.100569 [5] Guo Q B, Han S, Lu Y X, Chen L Q, Hu Y S 2023 Chin. Phys. Lett. 40 028801 doi: 10.1088/0256-307X/40/2/028801 [6] Jian Z L, Luo W, Ji X L 2015 J. Am. Chem. Soc. 137 11566 doi: 10.1021/jacs.5b06809 [7] Perveen T, Siddiq M, Shahzad N, Ihsan R, Ahmad A, Shahzad M I 2020 Renew. Sustain. Energy Rev. 119 109549 doi: 10.1016/j.rser.2019.109549 [8] Sha M, Liu L, Zhao H P, Lei Y 2020 Carbon Energy 2 350 doi: 10.1002/cey2.57 [9] Aslam M K, Niu Y, Xu M 2021 Adv. Energy Mater. 11 2000681 doi: 10.1002/aenm.202000681 [10] Wen Y, He K, Zhu Y J, Han F D, Xu Y H, Matsuda I, Ishii Y, Cumings J, Wang C S 2014 Nat. Commun. 5 4033 doi: 10.1038/ncomms5033 [11] Tan S H, Yang H, Zhang Z, Xu X Y, Xu Y Y, Zhou J, Zhou X C, Pan Z D, Rao X Y, Gu Y D, Wang Z L, Wu Y T, Liu X, Zhang Y 2023 Molecules 28 3134 doi: 10.3390/molecules28073134 [12] Lu X Y, Peng H D, Liu G P, Qi F Y, Shi C L, Wu S, Wu Y X, Yang H P, Shan J, Sun Z P 2023 Energy Adv. 2 1294 doi: 10.1039/D3YA00241A [13] Kuai J, Xie J, Wang J D, Chen J Y, Liu F, Xu X W, Tu J, Cheng J P 2024 Chem. Phys. Lett. 842 141214 doi: 10.1016/j.cplett.2024.141214 [14] Zhang K, He Q, Xiong F Y, Zhou J P, Zhao Y, Mai L Q, Zhang L N 2020 Nano Energy 77 105018 doi: 10.1016/j.nanoen.2020.105018 [15] Lei K X, Wang J, Chen C, Li S Y, Wang S W, Zheng S J, Li F J 2020 Rare Met. 39 989 doi: 10.1007/s12598-020-01463-9 [16] Feng Z Y, Peng W J, Wang Z X, Guo H J, Li X H, Yan G C, Wang J X 2021 Int. J. Miner. Metall. Mater. 28 1549 doi: 10.1007/s12613-021-2335-x [17] Garayt M D L, Zhang L B, Zhang Y X, Obialor M C, Deshmukh J, Xing Y J, Yang C Y, Metzger M, Dahn J R 2024 J. Electrochem. Soc. 171 070523 doi: 10.1149/1945-7111/ad5e64 [18] Lin J Y, Yu T, Han F J J, Yang G C 2020 Wires Comput. Mol. Sci. 10 e1473 doi: 10.1002/wcms.1473 [19] Kulish V V, Malyi O I, Persson C, Wu P 2015 Phys. Chem. Chem. Phys. 17 13921 doi: 10.1039/C5CP01502B [20] Hu J P, Xu B, Ouyang C Y, Yang S Y A, Yao Y G 2014 J. Phys. Chem. C 118 24274 doi: 10.1021/jp507336x [21] Xu Z M, Lv X J, Chen J G, Jiang L X, Lai Y Q, Li J 2017 Phys. Chem. Chem. Phys. 19 7807 doi: 10.1039/C7CP00064B [22] Yu Y D, Guo Z L, Peng Q, Zhou J, Sun Z M 2019 J. Mater. Chem. A 7 12145 doi: 10.1039/C9TA02650A [23] Yu T, Zhang S T, Li F, Zhao Z Y, Liu L L, Xu H Y, Yang G C 2017 J. Mater. Chem. A 5 18698 doi: 10.1039/C7TA04390B [24] 张改, 谢海妹, 宋海滨, 李晓菲, 张茜, 亢一澜 2022 物理学报 71 066501 doi: 10.7498/aps.71.20211405 Zhang G, Xie H M, Song H B, Li X F, Zhang Q, Kang Y L 2022 Acta Phys. Sin. 71 066501 doi: 10.7498/aps.71.20211405 [25] 雷雪玲, 朱巨湧, 柯强, 欧阳楚英 2024 物理学报 73 098804 doi: 10.7498/aps.73.20240197 Lei X L, Zhu J Y, Ke Q, Ouyang C Y 2024 Acta Phys. Sin. 73 098804 doi: 10.7498/aps.73.20240197 [26] Liang Y B, Liu Z, Wang J, Liu Y 2022 Chin. Phys. B 31 116302 doi: 10.1088/1674-1056/ac7859 [27] Aghamohammadi H, Hassanzadeh N, Eslami-Farsani R 2021 Ceram. Int. 47 22269 doi: 10.1016/j.ceramint.2021.05.048 [28] Mahmood N, Tang T, Hou Y 2016 Adv. Energy Mater. 6 1600374 doi: 10.1002/aenm.201600374 [29] Bi J X, Du Z Z, Sun J M, Liu Y H, Wang K, Du H F, Ai W, Huang W 2023 Adv. Mater. 35 2210734 doi: 10.1002/adma.202210734 [30] Fan X, Zheng W T, Kuo J L, Singh D J 2013 ACS Appl. Mater. Interfaces 5 7793 doi: 10.1021/am401548c [31] Liu M, Kutana A, Liu Y, Yakobson B I 2014 J. Phys. Chem. Lett. 5 1225 doi: 10.1021/jz500199d [32] Pollak E, Geng B, Jeon K J, Lucas I T, Richardson T J, Wang F, Kostecki R 2010 Nano Lett. 10 3386 doi: 10.1021/nl101223k [33] Ferrighi L, Trioni M I, Di Valentin C D 2015 J. Phys. Chem. C 119 6056 doi: 10.1021/jp512522m [34] Lee H, Paeng K, Kim I S 2018 Synth. Met. 244 36 doi: 10.1016/j.synthmet.2018.07.001 [35] Datta D, Li J, Shenoy V B 2014 ACS Appl. Mater. Interfaces 6 1788 doi: 10.1021/am404788e [36] Ma L B, Lv Y H, Wu J X, Xia C, Kang Q, Zhang Y Z, Liang H F, Jin Z 2021 Nano Res. 14 4442 doi: 10.1007/s12274-021-3439-3 [37] Share K, Cohn A P, Carter R, Rogers B, Pint C L 2016 ACS Nano 10 9738 doi: 10.1021/acsnano.6b05998 [38] Ma C C, Shao X H, Cao D P 2012 J. Mater. Chem. 22 8911 doi: 10.1039/c2jm00166g [39] Cai D D, Wang C S, Shi C Y, Tan N 2018 J. Alloys Compd. 731 235 doi: 10.1016/j.jallcom.2017.10.043 [40] Denis P A 2024 J. Mol. Model. 30 96 doi: 10.1007/s00894-024-05893-5 [41] Fei H L, Dong J C, Chen D L, Hu T D, Duan X D, Shakir I, Huang Y, Duan X F 2019 Chem. Soc. Rev. 48 5207 doi: 10.1039/C9CS00422J [42] Xiao R, Yu T, Yang S, Chen K, Li Z N, Liu Z B, Hu T Z, Hu G J, Li J, Cheng H M, Sun Z H, Li F 2022 Energy Storage Mater. 51 890 doi: 10.1016/j.ensm.2022.07.024 [43] Kresse G 1995 J. Non-Cryst. Solids 192–193 222 doi: 10.1016/0022-3093(95)00355-X [44] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169 doi: 10.1103/PhysRevB.54.11169 [45] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758 doi: 10.1103/PhysRevB.59.1758 [46] Blöchl P E 1994 Phys. Rev. B 50 17953 doi: 10.1103/PhysRevB.50.17953 [47] Perdew J P, Zunger A 1981 Phys. Rev. B 23 5048 doi: 10.1103/PhysRevB.23.5048 [48] Ceperley D M, Alder B J 1980 Phys. Rev. Lett. 45 566 doi: 10.1103/PhysRevLett.45.566 [49] Chadi D J 1977 Phys. Rev. B 16 1746 doi: 10.1103/PhysRevB.16.1746 [50] Togo A, Tanaka I 2015 Scr. Mater. 108 1 doi: 10.1016/j.scriptamat.2015.07.021 [51] Henkelman G, Uberuaga B P, Jónsson H 2000 J. Chem. Phys. 113 9901 doi: 10.1063/1.1329672 [52] Khan M I, Nadeem G, Majid A, Shakil M 2021 Mater. Sci. Eng. B 266 115061 doi: 10.1016/j.mseb.2021.115061 [53] Li Y F, Jiang J Z, Li X F, Li M, Zheng Y J, Sun K 2024 Phys. Rev. B 110 155401 doi: 10.1103/PhysRevB.110.155401 [54] Tarascon J M, Armand M 2001 Nature 414 359 doi: 10.1038/35104644 [55] Li P, Li Z Y, Yang J L 2018 J. Phys. Chem. Lett. 9 4852 doi: 10.1021/acs.jpclett.8b02035 [56] Lei S F, Chen X F, Xiao B B, Zhang W T, Liu J 2019 ACS Appl. Mater. Interfaces 11 28830 doi: 10.1021/acsami.9b07219 [57] Yang M R, Kong F, Chen L, Tian B W, Guo J 2023 Thin Solid Films 769 139734 doi: 10.1016/j.tsf.2023.139734 [58] Jiang H R, Shyy W, Liu M, Wei L, Wu M C, Zhao T S 2017 J. Mater. Chem. A 5 672 doi: 10.1039/C6TA09264K [59] Lin H, Liu G J, Zhu L L, Zhang Z J, Jin R C, Huang Y, Gao S M 2021 Appl. Surf. Sci. 544 148895 doi: 10.1016/j.apsusc.2020.148895 [60] Wang Y N, Li Y S 2020 J. Mater. Chem. A 8 4274 doi: 10.1039/C9TA11589G [61] Wang Y T, Zhou M, Xu L C, Zhao W T, Li R, Yang Z, Liu R P, Li X Y 2020 J. Power Sources 451 227791 doi: 10.1016/j.jpowsour.2020.227791 [62] Li Q F, Duan C G, Wan X G, Kuo J L 2015 J. Phys. Chem. C 119 8662 doi: 10.1021/jp512411g [63] Mukherjee S, Kavalsky L, Singh C V 2018 ACS Appl. Mater. Interfaces 10 8630 doi: 10.1021/acsami.7b18595 [64] Zhang X, Yu Z, Wang S S, Guan S, Yang H Y, Yao Y, Yang S A 2016 J. Mater. Chem. A 4 15224 doi: 10.1039/C6TA07065E [65] Ahmad S, Din H U, Nguyen C Q, Nguyen S T, Nguyen C 2024 Dalton. Trans. 53 3785 doi: 10.1039/D3DT03946C [66] Sannyal A, Zhang Z Q, Gao X F, Jang J 2018 Comput. Mater. Sci. 154 204 doi: 10.1016/j.commatsci.2018.08.002 [67] Liu M, Cheng Z S, Zhang X M, Li Y F, Jin L, Liu C, Dai X F, Liu Y, Wang X T, Liu G D 2023 Chin. Phys. B 32 096303 doi: 10.1088/1674-1056/acd623 [68] Fan K, Ying Y R, Li X, Luo X Y, Huang H T 2019 J. Phys. Chem. C 123 18207 doi: 10.1021/acs.jpcc.9b03963 [69] Wang Y, Liang S S, Tian J C, Duan H X, Lv Y, Wan L J, Huang C L, Wu M S, Ouyang C Y, Hu J P 2024 Phys. Chem. Chem. Phys. 26 4455 doi: 10.1039/D3CP05287G [70] Li F, Qu Y Y, Zhao M W 2016 J. Mater. Chem. A 4 8905 doi: 10.1039/C6TA03210A [71] Samad A, Shafique A, Shin Y H 2017 Nanotechnology 28 175401 doi: 10.1088/1361-6528/aa6536 [72] Yang Z F, Zheng Y P, Li W L, Zhang J P 2021 Nanoscale 13 11534 doi: 10.1039/D0NR07899A [73] Er D, Li J, Naguib M, Gogotsi Y, Shenoy V B 2014 ACS Appl. Mater. Interfaces 6 11173 doi: 10.1021/am501144q [74] Jing Y, Liu J, Zhou Z P, Zhang J, Li Y 2019 J. Phys. Chem. C 123 26803 doi: 10.1021/acs.jpcc.9b07950 [75] Hu J P, Xu B, Ouyang C Y, Zhang Y, Yang S A 2016 RSC Adv. 6 27467 doi: 10.1039/C5RA25028E [76] Wang D S, Liu Y H, Meng X, Wei Y J, Zhao Y Y, Pang Q, Chen G 2017 J. Mater. Chem. A 5 21370 doi: 10.1039/C7TA06944H [77] Putungan D B, Lin S H, Kuo J L 2016 ACS Appl. Mater. Interfaces 8 18754 doi: 10.1021/acsami.6b03499 -
计量
- 文章访问数: 588
- HTML全文浏览数: 588
- PDF下载数: 9
- 施引文献: 0