
 首页
首页 登录
登录 注册
注册





HTML
-
Recently, all-nitrogen salts have drawn much attention in the high-energy-density materials (HEDMs) field due to their potential as ideal candidates in propellant or explosive applications.[1] Generally, the high energy of these compounds is primarily attributed to the substantial disparity in bond energy between N–N or N=N bonds in nonmolecular nitrogen (160/418 kJ/mol) and the N≡N triple bond in N2 molecule (954 kJ/mol). In the past decade, several nitrogen-rich metal compounds with high energy density have been reported, including Be–N,[2–6] Mg–N,[7,8] Al–N,[9–11] S–N,[12] Fe–N,[13–15] Cu–N,[16,17] Zn–N,[18–20] Se–N,[21] Hg–N,[22] Ta–N,[23] W–N,[24] Y–N,[25] Ne–N,[26] and others. Theoretical calculations and experimental synthesis have demonstrated the existence of diverse all-nitrogen ions or clusters ranging from N3 to N13.[27,28] However, the successful preparation of such compounds remains rare. In 2001, Christe’s work demonstrated the stability of
Numerous planar or quasiplanar cyclo-N6 anions have been predicted in Li, Mg, Ca, and Ba nitrides under high pressure.[7,35–37] However, they have not paid close attention to the microstructural characteristics of these cyclo-N6 anions. It’s worth noting that an in-plane distortion may occur in the cyclo-N6 sub-lattices when the neutral π-aromatic system transforms into a charged anion. For instance, chair-like N6 anions have been predicted in h-WN6, TeN6, and GdN6,[38–40] where strong chemical bonding and charge transfers are believed to contribute to the structural stability. The synthesis of WN6, as mentioned in Ref. [24], has led to an increased interest in the study of N6 anions. However, it is important to note that the content of nitrogen in these compounds will be decreased, resulting in relatively lower energy density due to the larger molar mass of tungsten (W) and tellurium (Te). In a separate study conducted by Liu et al.,[41] they reported the existence of GaN5 and GaN6 compounds with high nitrogen contents. It is worth mentioning that gallium (Ga) and aluminum (Al) are elements belonging to the same group. Generally, aluminum has a small atomic radius, high valence electron density, and high electronegativity, which makes it capable of forming strong covalent bonds with nitrogen atoms. Meanwhile, the relatively lower molar mass of aluminum compared to Te, W, Gd, and Ga allows for higher nitrogen content in aluminum-based compounds. Moreover, Cui et al. have predicted the existence of a notable monoclinic phase with polymeric nitrogen chains (P21/c-AlN3) for AlN3.[9] This phase is stable within a specific pressure range of 43–85 GPa, which indicates that aluminum and nitrogen can indeed form nitrogen-rich compounds under high pressure. Based on these factors, it is reasonable to consider binary Al–N compounds as potential candidates for the search of N6 anions.
In the present study, the R-3m-AlN3 phase has been successfully designed, featuring a chair-like
-
The structure prediction for the Al–N system was carried out using the particle swarm optimization (PSO) methodology implemented in the CALYPSO code.[42,43] This approach does not rely on any known structural information and has been successfully validated in various systems under high pressure, including binary and ternary systems.[44–50] For the structural optimization and electronic properties calculations, density functional theory (DFT) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional and generalized gradient approximation (GGA) was employed.[51] These calculations were performed utilizing the VASP code.[52] The all-electron projector augmented wave (PAW)[53] method was adopted to represent the ionic potentials, the valence electrons of Al and N atoms are 3s23p1 and 2s22p3, respectively. The plane-wave basis set cutoff (700 eV) and appropriate Monkhorst–Pack k-meshes[54] (2π × 0.03 Å−1) were used to ensure all enthalpy calculations converged less than 1 meV/atom. In addition, phonon dispersion curves were computed using the supercell method implemented in the PHONOPY code[55] to determine the dynamical stability. FPMD simulations were performed in the NVT ensemble from 300 K to 700 K through the Nosé–Hoover method[56,57] to assess the thermodynamic stability of the system at different temperatures. We calculated the elastic constants employing the strain–stress method[58] as adopted in the VASP code. Relative elastic property parameters were determined using the Voigt–Reuss–Hill approximations.[59] The chemical bonding mode of AlN3 was executed by the solid state adaptive natural density partitioning (SSAdNDP) method.[60] Orbital calculations were carried out at the B3LYP/6-311+g(d) level of theory using the Gaussian 09 code.[61] Bader analysis[62] was utilized to determine the transfer of charge within the system. The crystal structure patterns were visualized through VESTA[63] software.
-
We conducted a comprehensive search for all potential structures within the AlN3 system across a pressure range from 0 to 100 GPa. Subsequently, we identified a crystal structure that features an N6 ring and proceeded to optimize these structures. The total energy of structures containing N6 rings was calculated under ambient conditions. The results indicated that the structure with the R-3m space group exhibited the lowest energy among all the selected structures at 0 GPa, as shown in Fig. 1(a). Furthermore, it was observed that structures with chair and boat N6 rings generally had lower energies compared to structures with planar N6 rings in AlN3. Simultaneously, the energy of structures with chair N6 rings was generally lower than that with boat N6 rings. This disparity arises since the bond angles between nitrogen atoms in chair N6 rings tend to align more closely with the ideal angle (120°) in contrast to boat N6 rings. This alignment reduction minimizes the electron cloud overlap and Coulomb repulsion, consequently elevating the energy level of the conformation. Additionally, a comparison was made between the energy of P21/c -AlN3 and R-3m-AlN3 in the pressure range from 0 to 100 GPa (
Fig. S1 ). The findings revealed that the P21/c structure had lower energy compared to the R-3m structure. However, recent high-pressure experiments have encouraged us to continue the research on R-3m-AlN3. It is worth noting that the recently synthesized bp-N is not the lowest in energy,[64,65] and materials like silicon and ice can form different phases under varying pressure–temperature paths.[66,67] This suggests that the phases with higher energy can be formed by controlling the synthesis conditions.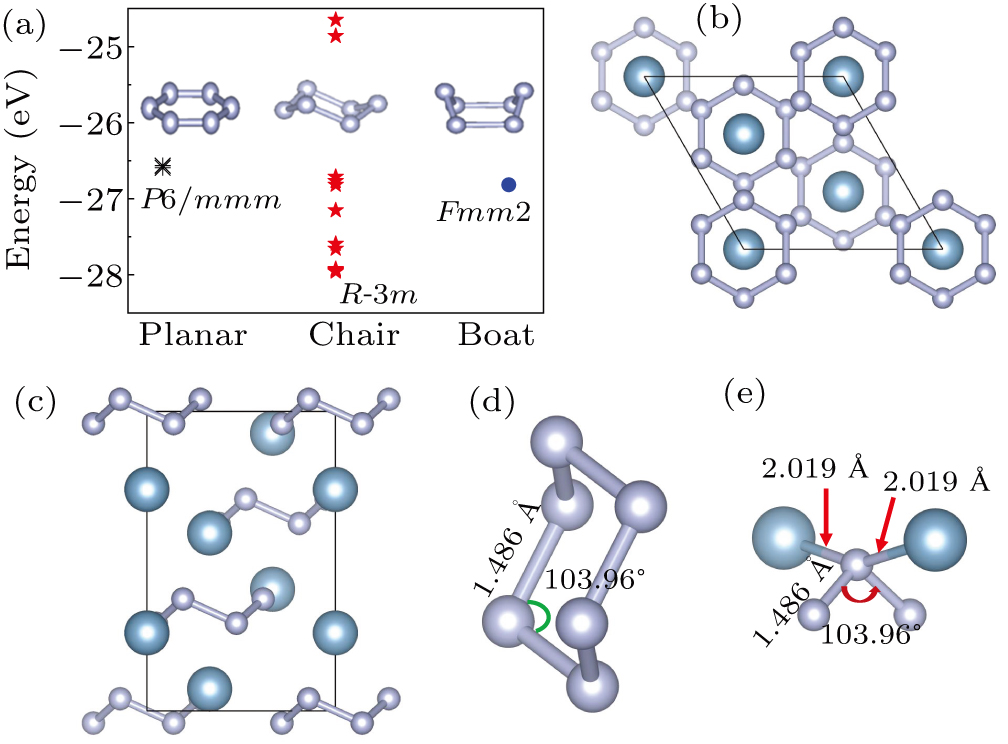
The R-3m-AlN3 phase (Figs. 1(b) and 1(c)) and the separate chaired N6 ring structure (Fig. 1(d)) are illustrated. Under atmospheric conditions, the bond length and bond angle for the chaired N6 ring structure are equal, measuring 1.486 Å and 103.96°, respectively. In the R-3m-AlN3 phase, six N atoms form an N6 ring network, and each Al atom is bonded to six N atoms, forming an octahedral structure with a sixfold coordinated mode. Each N atom is driven by the sp3 hybridization of two adjacent N atoms and Al atoms, forming four covalent bonds. The Al–N distances in the structure are 2.019 Å and 2.149 Å, respectively (Fig. 1(e)). The detailed information on the structure is summarized in Table 1.
-
To assess the kinetics stability of the R-3m-AlN3 structure at 0 GPa, phonon spectra were computed using the supercell method. In general, the presence of imaginary frequencies in the phonon spectra typically suggests the potential for vibration mode instability. Conversely, the absence of imaginary frequencies indicates a lack of vibration modes that could lead to structural instability. This also implies that the structure remains stable across a range of vibration modes. As shown in Figs. 2(a) and 2(b), the absence of imaginary frequency in the over Brillouin zone demonstrates that the structure is kinetically stable. Based on the analysis of phonon density of states (PHDOS) for the R-3m-AlN3 structure, it was observed that there are strongly coupled vibrations between the Al and N atoms, which contribute to the low-frequency vibration modes. Furthermore, the stretching mode of the N–N bond induces high-frequency vibrations.
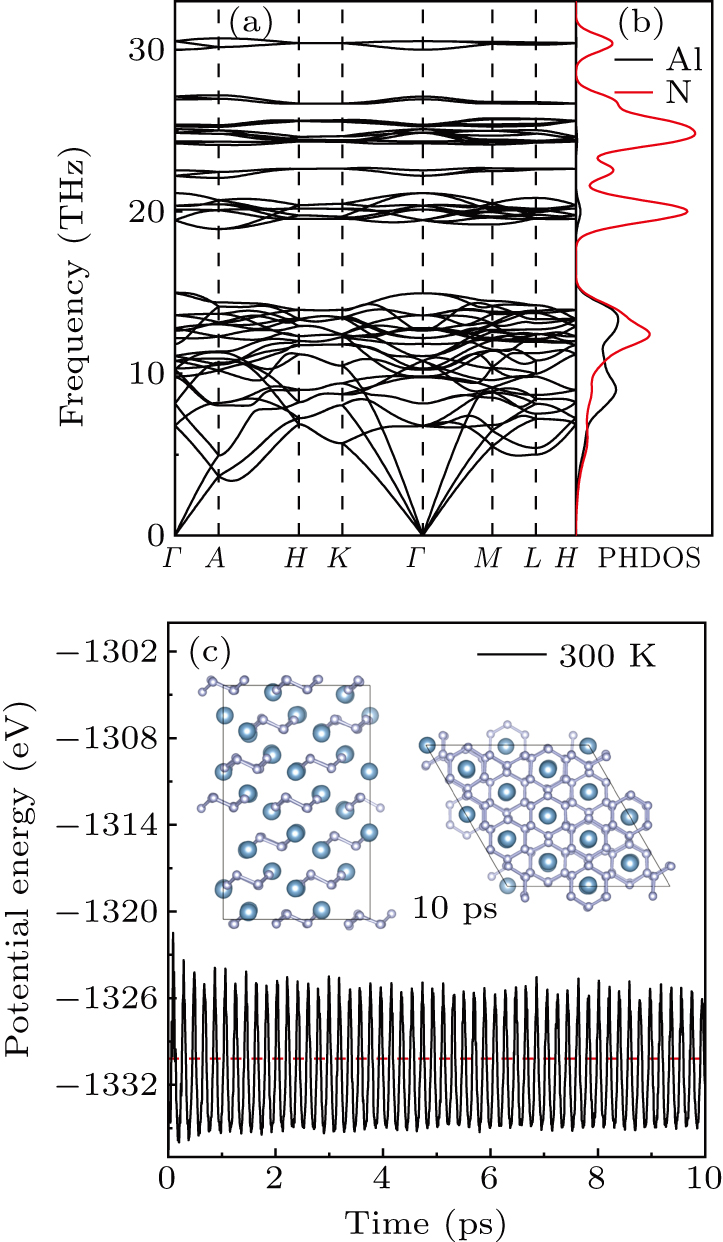
Similarly, the thermal stability of the
Figures S2 and S3 depict the temperature profiles at 500 K and 700 K, using a 1 fs time step. It is discovered that the N–N bond is broken at the 700 K temperature, leading to the disruption of the integrity of the N6 ring. The plots of radial distribution function (RDF) for N–N separations are shown inFigs. S4 and S5 . The first sharp peak in the RDF represents the nearest distance between the selected atom and its nearest neighboring atoms. It is shown that there is a single RDF peak at approximately 1.488 Å corresponding to the N–N distance without significant change at 300 K and 500 K, which supports the conclusion that the R-3m-AlN3 with -
It is widely known that the mechanical properties of any crystal are strongly influenced by its fundamental elastic constants (Cij). To verify the mechanical properties of the R-3m-AlN3 structure at 0 GPa, the elastic constants were calculated using the strain–stress method.[58] The R-3m phase requires the determination of six independent elastic constants, and the Born–Huang criterion[68] was employed to verify the mechanical stability of the R-3m-AlN3 structure at 0 GPa. The mechanical stability criteria for the hexagonal phase are evaluated based on the following conditions: C44 > 0,
It can be seen from Table 2, that the R-3m-AlN3 phase exhibits a shear modulus of 133 GPa and a bulk modulus of 185 GPa. The Pugh criterion serves as an empirical approach used to predict the brittle or ductile behavior of crystalline materials. Importantly, it has been successfully applied to anticipate various material-related characteristics, and the outcomes of its predictions have gained recognition within the scientific community. According to Pugh’s criterion,[69] the ratio of k = G/B determines whether the materials behave in a brittle or ductile manner. If k > 0.57, the material behaves in a brittle manner; otherwise, it is ductile. In the case of R-3m-AlN3, the calculated value of G/B (0.72) is larger than 0.57, similar to the diamond (1.10) and c-BN (0.86),[70] meaning the brittle materials. Additionally, the stiffness of a material can be characterized by Young’s modulus,[71] the high value (322 GPa) indicates that the R-3m-AlN3 structure can be used as a potential hard material. Poisson’s ratio reflects the degree of covalent bonding in a material.[72] The value (0.21) is lower than 0.33, indicating a strong degree of covalent bonding. Taken together, these excellent mechanical parameters demonstrate the good mechanical stability of the R-3m-AlN3 structure.
-
The electronic band structure and projected density of states (PDOS) of the R-3m-AlN3 phase at 0 GPa were calculated to explore its inner nature and formation mechanism, as shown in Fig. 3. The calculations reveal that the R-3m-AlN3 structure possesses semiconductor property with a large band gap of 4.14 eV. This suggests its potential for applications in electronic devices and optoelectronics. However, density functional calculations usually lead to a significant underestimation of the energy gap due to several primary factors, such as the approximation of exchange–correlation functionals, self-interaction errors, the size of the basis set, etc. Therefore, the actual band gap is expected to be greater than 4.14 eV. In addition, the electronic band structure shows a clear overlap between the Al 3p and N 2p orbitals, demonstrating the formation of a strong chemical bonding as depicted in Figs. 3(b) and 3(c). To gain further insights into the behavior of the chemical bonds, Bader[62] charge analysis (shown in
Table S1 ) was performed. The analysis revealed that there is a significant electron transfer (2.4e) between aluminum and nitrogen atoms. This information provides insights into the behavior of chemical bonds within the R-3m-AlN3 structure.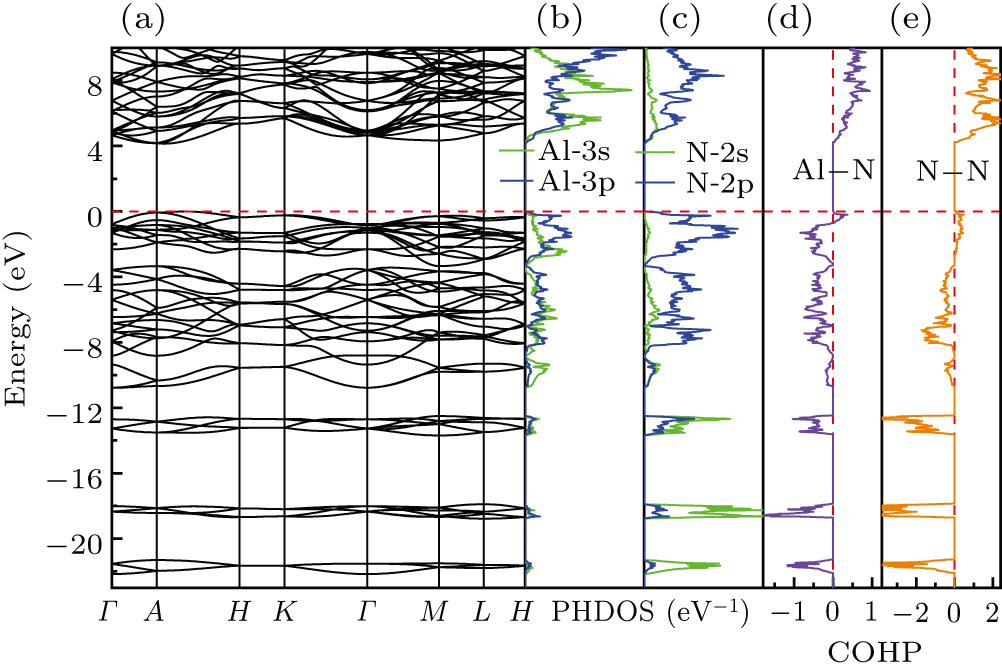
The crystal orbital Hamiltonian group (COHP) is a target of energy resolution to measure the overlap strength and crystal orbital overlap population, which helps to understand the contribution of atomic pairs to the structural stability determining the bonding information based on the COHP in the LOBSTER program.[73] Generally speaking, the positive and negative COHP values indicate the presence of antibonding and bonding states, respectively. The COHP plot of the Al–N pairs and N–N pairs are shown in Figs. 3(d) and 3(e), respectively. It is obvious that bonding states are fully occupied, and the antibonding states are not occupied, which demonstrates the strong covalent bond feature between them. In addition, the ICOHP values quantify the strength of the antibonding interaction. In this case, the ICOHP values corresponding to Figs. 3(d) and 3(e) are −4.49 and −9.18, respectively. These results reflect the strong covalent bonding characteristics between Al and N atoms in the Al–N pairs and the strong covalent bonding between neighboring N atoms in the N–N pairs.
-
To illustrate the reason for the high stability of the R-3m-AlN3 phase, the SSAdNDP method was used to analyze its chemical bonding patterns, relative results of the analysis are plotted in Fig. 4. In the primitive cell, there are 36 2c-2e Al–N σ bonds (Fig. 4(a)) and 18 2c-2e N–N σ bonds (Fig. 4(c)), the total number of electrons involved in these bonds is 108, which is in complete agreement with the sum of outer valence electrons in the AlN3 unit cell. It is worth noting that one unit cell comprises six AlN3 formulas, with Al and N elements contributing 3s23p1 and 2s22p3, respectively. The reasonable occupation numbers of these bonds indicate that the computational results are reliable. Moreover, the bonding modes of Al–N and N–N in the R-3m-AlN3 phase are illustrated in Figs. 4(b) and 4(d), respectively. For the
S6 .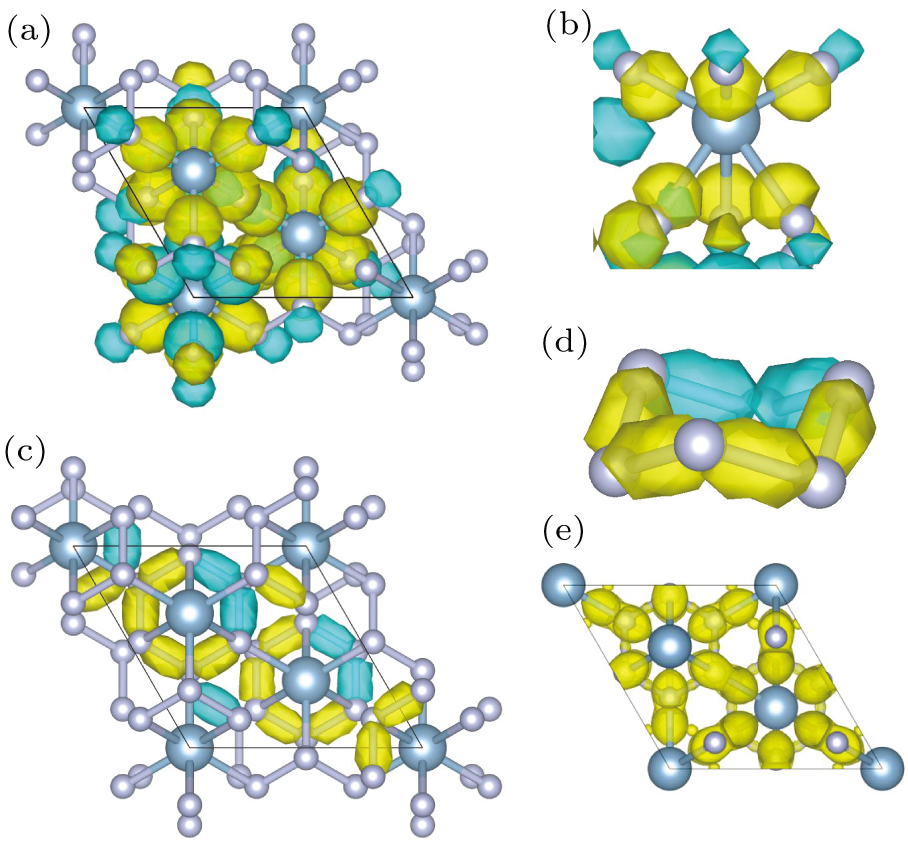
To demonstrate the bonding situation, Fig. 5 displays a scheme of the molecular orbital diagram of the
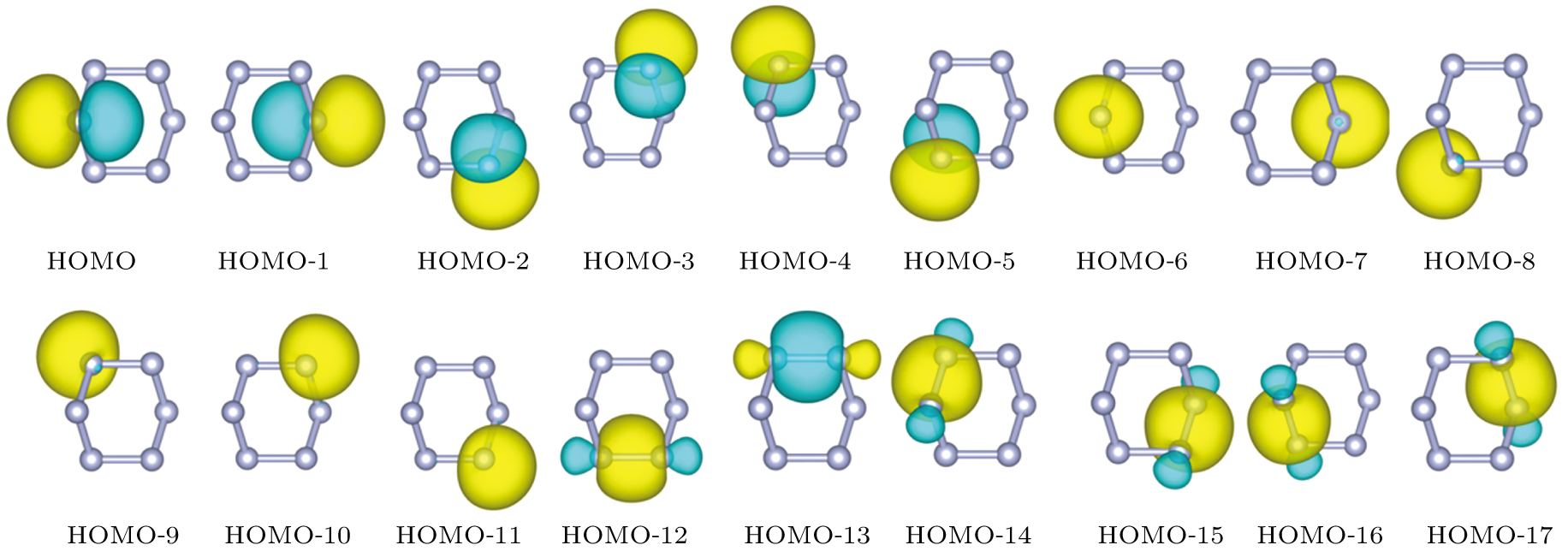
-
Indeed, the presence of Al atoms significantly enhances the stability of
Subsequently, we investigated the formation mechanism of Al–N covalent bonds by analyzing the relationship between Al–N distances and the ever-increasing pressure, as depicted in Fig. 6(a). Our calculations revealed that the distances between Al and N atoms gradually decrease as the pressure increases. Notably, the Al–N distance reaches the sum (1.96 Å) of the covalent radii[76] of Al 3p and N 2p at a pressure of 20 GPa. Moreover, it is worth mentioning that throughout the pressure range studied, the Al–N distance remains larger than the N–N distance in the case of cg-N. This result provides strong evidence supporting the covalent bonding nature of the Al–N bonds.
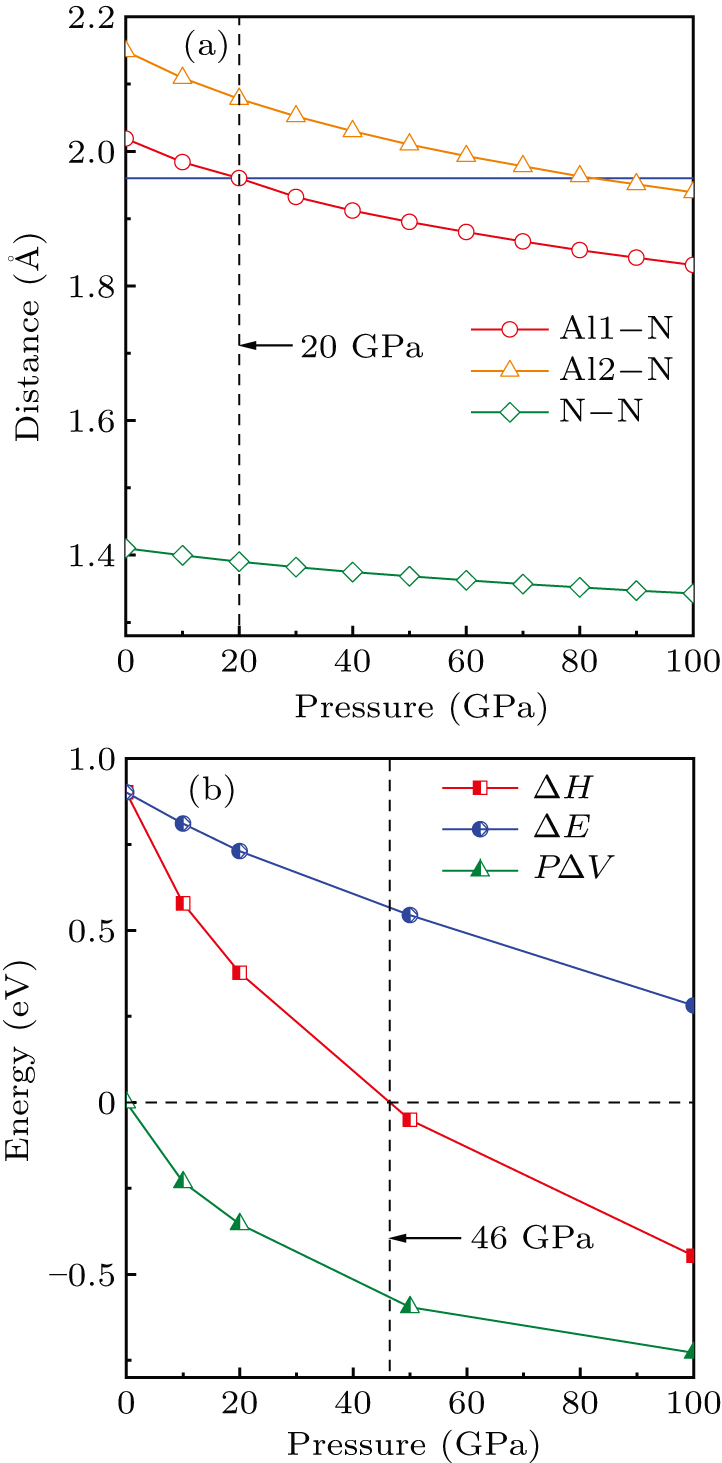
To illustrate the decomposition of AlN3 into AlN and N2 at high pressure, we plot the enthalpy difference (ΔH) between AlN3 and AlN + N2. Additionally, we analyze the contributions to ΔH from the internal energy (ΔE) and volume term (PΔV), as shown in Fig. 6(b). It reveals that the decomposition of AlN3 into AlN and N2 occurs spontaneously at pressures higher than 46 GPa. Furthermore, the competition between the ΔE and PΔV terms is crucial in determining the actual chemical reaction. Although ΔE remains largely positive, the PΔV term is negative due to the associated volume reduction, and this effect becomes more substantially reduced as the pressure increases. At 46 GPa, the PΔV term dominates the competition, leading to a negative ΔH value and the formation of the stable AlN3 compound.
-
The energy density is a crucial parameter for evaluating the detonation properties of a material. Here, in the case of the predicted stable phase of R-3m-AlN3, a significant amount of chemical energy is released when it dissociates into stable AlN solid and dinitrogen gas under ambient conditions. Considering the energy reduction of nitrogen gas concerning the α nitrogen phase (around 0.25 eV/atom),[80] the chemical energy densities (Ed) of dense R-3m-AlN3 with a density of 3.45 g/cm3 can be calculated to be approximately 5.04 kJ/g (Table 3). This calculation is based on the dissociation of AlN3 (s) → AlN (s) + N2 (g) and referencing to the energy of their most stable phase at 0 GPa.[81,82] Strikingly, the performance of R-3m-AlN3 in terms of energy density surpasses that of the typical explosive material TNT (4.3 kJ/g).[77] Furthermore, the volumetric energy densities (Ev) of R-3m-AlN3 are estimated to be 17.39 kJ/cm3, which is approximately 2.5 times that of TNT (∼7.05 kJ/cm3) and 1.6 times that of HMX (∼10.83 kJ/cm3).
The detonation properties of the R-3m-AlN3 structure, including detonation velocities (Vd) and detonation pressures (Pd), are important indicators for evaluating its suitability as a HEDM.[83] The empirical Kamlet–Jacobs equations[79] provide a way to estimate Vd and Pd based on certain parameters. The equations for Vd and Pd are given as follows:
-
In the conducted research, an experimental synthesis of aluminum nitride (AlN) in the P63mc phase (wurtzite structure) has been successfully achieved and found to be stable within the pressure range of 0–20 GPa.[81] Here, to explore the feasibility of synthesizing R-3m-AlN3 under high pressure, the formation enthalpy of AlN3 relative to AlN and N2 (Fig. 7) has been calculated. Additionally, the phase transition process of AlN and N is shown in
Figs. S7 and S8 , respectively. The result shows that the R-3m-AlN3 exhibits lower energy compared to AlN and N2 under pressures exceeding 46 GPa. This suggests that the synthesis of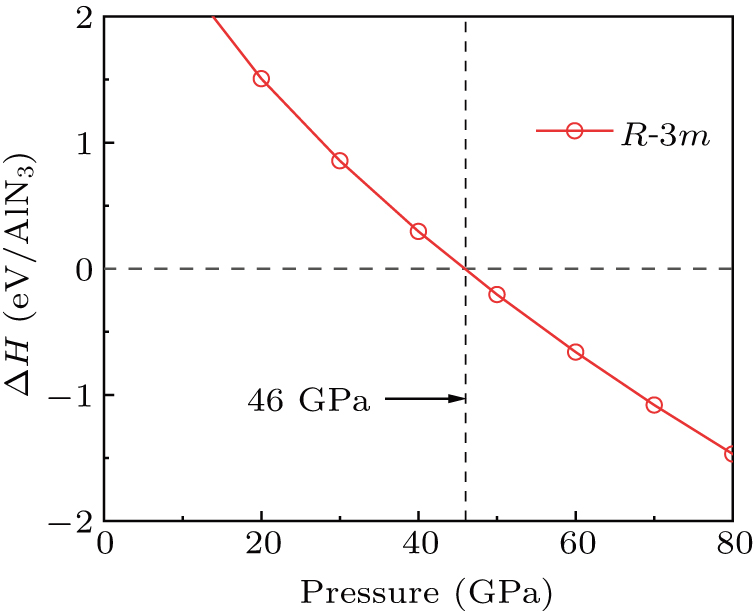
3.1. Crystal structure
3.2. Kinetic and thermal stability
3.3. Mechanical stability
3.4. Electronic properties
3.5. Chemical bonding pattern
3.6. Stabilization mechanism
3.7. Explosive performance
3.8.
Route to chair-like N 6 6 −
-
Through swarm-intelligence structure searches, the presence of all-nitrogen anions
 DownLoad:
DownLoad: