
 首页
首页 登录
登录 注册
注册



-
随着科学理论、算法及计算能力的发展,计算已经与实验和理论并列,成为重要的研究手段。量子力学建立之后,原则上求解基于量子理论的薛定谔方程即可获得材料几乎所有性质。然而由于直接求解需要巨大计算量,许多感兴趣的体系和性质无法计算。20世纪60年代密度泛函理论(Density Functional Theory,DFT)的提出及应用改变了这一情况。然而,DFT大多用于研究材料的基态性质,对于材料激发态性质以及在外场下的响应,含时密度泛函理论(Time-Dependent Density Functional Theory,TDDFT)是研究这类问题的计算效率较高的理论方法。本文简要回顾了TDDFT的历史和一些基本问题,着重介绍近年来TDDFT在理论和应用方面的重要进展以及目前仍然存在的问题,最后展望TDDFT未来的发展趋势。本文试图对TDDFT的基本问题提供快速的指南,希望能帮助感兴趣的研究人员更快地了解该领域的重要进展,以期达到与相关研究相互促进的目的。
-
原则上量子力学可以给出材料的几乎所有性质,然而对于包含大量原子的多体体系,如块材或大分子,直接求解薛定谔方程的计算量非常巨大,许多情况几乎不可能完成。也无怪乎狄拉克在量子力学发展初期说出“唯一的困难在于应用这些定律会导致非常复杂以至于无法求解的方程组”[1]。基于波函数的方法,如早期的Hartree-Fock方法,以及后来发展的Møller-Plesset(MP)微扰法、组态相互作用(CI)等post-Hartree-Fock方法,都存在计算量巨大的缺点。这一情况随着1964年、1965年DFT的基础—Hohenberg-Kohn定理[2]及其便于实用的Kohn-Sham方程[3]的提出和应用发生了巨大的改变,DFT相比其先驱者Thomas-Fermi模型和
${X_\alpha }$ 方法,有着严格的理论基础,精度大为提高,因此在物理、化学及材料领域得到了非常广泛的应用。目前,DFT仍在迅速发展,更好的交换关联泛函不断提出,推动其计算准确性不断提高。DFT是基于基态电荷密度,大多用来计算基态的相关性质,然而人们关心的很大一部分问题涉及材料的激发态性质或者材料在外加时变场下的性质变化,需要高效的方法处理体系的激发态相关问题。对于一般体系而言,直接求解含时薛定谔方程同样是不可能完成的任务,TDDFT的出现和发展,使大体系含时性质的高精度计算成为可能,有力地推动了计算手段在该领域的应用。 -
TDDFT理论的完整建立相比DFT晚不少。不过与DFT相似,TDDFT的起源可追溯到Bloch于1933年提出的含时Thomas-Fermi模型[4]。随后Peuckert[5]、Zangwill和Soven[6]对得到含时Kohn-Sham方程进行了有益的尝试,把稀有气体原子在时变外场下密度的线性响应等效地用无相互作用电子在等效时变场的响应来处理,并提出了含时交换关联的处理方案。在TDDFT基础方面,Ghosh和Deb[7–10]以及Bartolotti[11–14]探索了含时密度情形对应的Hohenberg-Kohn定理和Kohn-Sham方法,但是其推导过程都对时变场有较强的限制,例如前者要求时变场在时域上是周期的,后者要求是绝热过程。含时密度情形的Hohenberg-Kohn定理和Kohn-Sham方法的一般性证明直到1984年才被Runge和Gross[15]实现。
对于含时情形,人们希望有与Hohenberg-Kohn定理对应的含时密度与外势场的一一对应关系。然而,与基态的DFT相比,含时情况有两个重要的不同。首先,含时情形不像基态一样能量最小,因此没有像基态一样可以利用的Rayleigh-Ritz变分法则。与基态情形不同,Runge和Gross的推导基于最基本的含时薛定谔方程
式中:
$\psi\left({{r}}_{1}, \cdots, {{r}}_{N}, t\right)$ 为N电子体系在t时刻的多体波函数,${\psi _0}$ 为体系在初始时刻t0的多体波函数,ri代表电子i的自由度,t代表时间,$ \hat H $ 为体系哈密顿量。另外,含时薛定谔方程是一个初值问题,所有证明都需要考虑一个给定的初始态${\psi _0}$ 。Runge和Gross[15]证明了对于两个有着相同电子数目N的多体系统而言,如果其初始态相同,相应的两个外部标量势场在初始时刻t0附近都是可Taylor展开的,那么不同的时变外势场v和v′(此处势场不同的含义为v(r, t)-v′(r, t)≠c(t))一定给出不同的含时电荷密度。也就是说,初始状态相同时,若含时电荷密度相同,则时变外势场必然相同。由此确定了含时电荷密度与时变外场的对应关系。他们的证明分两步进行:第一步,确定时变外势场v(r, t)与含时电流密度j(r, t)的对应关系;第二步,确定含时电流密度j(r, t)与含时电荷密度n(r, t)的对应关系。从中亦可看出j(r, t)的重要地位。
一旦确定了含时情形的一一对应关系,则与基态的DFT一样,可以将时变外势场写作含时电荷密度的函数,即
式中:v为外势场,n为电荷密度,
${\psi_0}$ 为体系初始波函数。需要注意的是,势场亦依赖于初始状态。当然,当初始状态为基态时,由Hohenberg-Kohn定理,基态由基态电荷密度决定,此时外势场完全由含时电荷密度决定。更进一步,与基态情形一样,体系哈密顿量以及所有的含时可观测量都可写成含时电荷密度的泛函。 -
有Runge-Gross定理作为理论支撑,在实际应用中很自然地希望有类似基态情形的含时Kohn-Sham方程,即借助一个无相互作用参考体系随时间演化问题的求解得到实际相互作用体系随时间演化特性。含时Kohn-Sham方程的存在和合理性并不能直接由Runge-Gross定理得出,同时与基态类似,也面临着v-representability问题。所谓v-representability问题,即给定一个具有良好定义(Well-Defined)的电荷密度函数(如满足非负,积分等于体系电子数目),是否一定存在一个外势场使得可以得到该电荷密度函数。对于基态情形,如果电荷密度函数在基态电荷密度附近不满足v-representability,那么对电荷密度函数的泛函微分是无法定义的,不能应用变分法则对体系进行求解。而对于含时和基态情形,如果对于参考的无相互作用体系不满足v-representability,则Kohn-Sham方法从根本上就无法成立。这一问题的解决并不显而易见,长久以来在DFT和TDDFT领域都有着持续的研究。对DFT而言,再次借助于基态能量最小,人们幸运地通过“Constrained Search”方式处理v-representability问题,使得对任意具有良好定义的电荷密度函数对应的泛函都有合理的定义[16–17];对于TDDFT而言,情况就复杂得多,不少研究组对此进行研究,其中van Leeuwen等[18]做出了重要贡献。
van Leeuwen定理指出:对于两个有着不同相互作用w(r-r′)和w′(r-r′)的多体体系,如果相关的电荷密度关于时间t可Taylor展开,体系1在外势场v(r, t)作用下的含时电荷密度为n(r, t),则在体系2中一定可以唯一地构造出外势场v′(r, t),使其含时电荷密度与体系1完全相同。van Leeuwen定理有两个重要的特例:(1)当w(r-r′) = w′(r-r′)时,两个体系相同,根据外势场构造的唯一性,从另一种途径证明了Runge-Gross定理;(2)当w′(r-r′)≡0时,体系2为非相互作用体系,van Leeuwen定理确定了非相互作用体系的v-representability,为含时Kohn-Sham方程提供了坚实的理论基础。需要说明,van Leeuwen定理依然存在不少限制,例如一些体系的电荷密度无法对时间t进行Taylor展开,此时van Leeuwen定理不成立,但是Runge-Gross定理是成立的,不过Runge-Gross定理依然要求外势场可Taylor展开。因此拓展Runge-Gross定理或通过别的途径确定TDDFT的理论基础依然需要长期的努力。近年来人们对此取得了一些新进展,如不动点证明[19–20]。
以van Leeuwen定理等作为理论基础,可以得到类似于基态情形的含时Kohn-Sham方程,其形式看上去也和基态时类似
式中:
${\varphi _j}({ r},{ t})$ 为对应的无相互作用参考系统的含时Kohn-Sham轨道,vs为Kohn-Sham势,v为外势场,vH(r, t)是由瞬时电荷密度n(r, t)决定的经典库仑势,vxc为含时的交换关联势,$\psi_{0}$ 为真实相互作用体系的初始多体波函数,$\varPhi_{0}$ 为Kohn-Sham参考体系的初始波函数。含时Kohn-Sham方程给出了与真实系统相同的含时电荷密度。严格来说,t时刻的vxc是当前及历史电荷密度(n(r, t′), t′≤t)的泛函,并且它依赖于真实相互作用体系的初始多体波函数${\psi _0}$ 和Kohn-Sham参考体系的初始波函数${\varPhi _0}$ ,这些与基态情形有很大不同。同时它无需像基态情况一样表示为交换关联能的泛函微分。然而,它也面临着与基态一样的问题:无法确定泛函的精确形式,实际应用时仍然依赖于对含时交换关联势的近似。 -
在实际含时问题中,有一类常见而重要的情形:系统所受外场作用较小,由此对初始态的偏离也较小,大多数谱学应用(通过考察体系对小外场响应得到体系的相关谱性质)就属于此类情况。由于该类问题的重要性,长期以来人们对TDDFT在该领域的应用不断深入。
在外场较弱的情况下,没有必要对含时薛定谔方程进行完全求解以得到所需性质,往往通过微扰理论达到目的。其中重要的一类是线性响应理论,即忽略波函数的变化,仅在微扰理论的一阶程度上考虑感兴趣的可观测量,这对解决很多问题都够用,特别对于电荷密度响应,TDDFT具有很重要的应用。以下先简要介绍一般性体系的电荷密度对外场的线性响应,再引出TDDFT的相关理论及应用。
考虑初始时刻为基态的体系,在外场作用下体系的电荷密度随时间变化,含时电荷密度可以按照微扰阶次展开为
式中:n0为基态电荷密度;n1为电荷密度变化的一阶项,即线性响应项;n2为电荷密度变化的二阶项。当外场较弱时,线性响应项比高阶响应项大得多,是密度变化的主要贡献,可主要考虑该项;当外场较强时,高阶项的贡献很大,微扰理论不再成立。
电荷密度的线性响应n1可通过下式求出
式中:v1为外势场;
$\chi $ (r, r′, t–t′)为所谓的密度-密度响应函数,定义为[21–23]式中:
$\theta $ 为阶跃函数,$\hat n $ 为电荷密度,$\psi_{\rm{gs}}$ 为基态波函数。由DFT,$\chi $ 是基态电荷密度的泛函$\chi \left[ {{n_0}} \right]$ 。一般情况下,相比时域响应,人们更感兴趣的是频域响应,即通过傅里叶变换,得到密度-密度响应函数
$\chi $ 的Lehmann表示式中:
$\psi_{n}$ 为标号为n的本征态波函数;${\varOmega _n} = {E_n} - {E_0}$ ,为多体体系的第n激发能。通过Lehmann表示可以清楚地看到响应函数$\chi $ 在体系的激发能处有极点。如果微扰外场${v_1}({r},\omega )$ 的频率与体系的某个激发能相匹配,则体系的响应会非常大,这便是谱峰。通过线性响应理论,在得到响应函数
$\chi $ 之后即可求出密度响应,进而求出感兴趣的谱。在TDDFT中,也有相应方法通过求解无相互作用的Kohn-Sham参考体系的相关量得到真实相互作用体系的线性响应。此时密度的线性响应可由Kohn-Sham系统的响应函数${\chi _{\rm{s}}}$ 和有效微扰v1s得到[24]有效微扰v1s除了包含实际的外场微扰项之外,还有附加项
式中:fxc为所谓的交换关联核,定义为含时交换关联势对含时密度在基态密度处的泛函微分
同样人们对频域的响应更感兴趣,对应的交换关联核为
${f_{{\rm{xc}}}}\left( {{r},{{r}^\prime },\omega } \right)$ 。频域的Kohn-Sham响应函数${\chi _{\rm{s}}}$ 也有类似的Lehmann表示式中:fj(k)为Kohn-Sham基态轨道j(k)的占据数;
${\omega _{jk}} = {\varepsilon _j} - {\varepsilon _k}$ ,为两轨道的能级差。可见,Kohn-Sham响应函数的极点即为无相互作用的Kohn-Sham参考体系的激发能。这看上去与真实的相互作用体系不同。实际上,由有效微扰v1s的表达式可以看出,含时密度泛函Kohn-Sham线性响应是一个自洽方程;而在自洽求解之后,错误的极点会被抵消掉,最后恢复真实体系的极点结构。含时密度泛函线性响应除了可以用来计算各种谱学性质,还可以用于研究体系的激发能。激发能定义为激发态能量与基态能量之差,原则上只需要计算这两个定态能量就可以求出。不过从另一个角度来考虑,把激发看作体系在两个本征态(基态和激发态)之间转变的动态过程,此时激发能对应该过程的特征频率,也就是说每个激发都对应该相互作用多体系统的一个本征模式。
从含时密度泛函出发计算激发能,对应微扰外场
${v_1}({r},\omega )=0$ 的情形。但有效微扰v1s并不为零,它包含Hartree项和交换关联核项,对应Hartree-xc核,即${f_{{\rm{Hxc}}}}\left( {{r},{{r}^\prime },\omega } \right) = {\left| {{r} - {{r}^\prime }} \right|^{ - 1}} + {f_{{\rm{xc}}}}\left( {{r},{{r}^\prime },\omega } \right)$ ,具体表述为(13)式是一个频率相关的积分算符作用在电荷密度
${n_{1\sigma }}({r},\omega )$ 上的本征值方程,对所有频率都有一阶电荷响应n1 = 0的平凡解。同时,对于某些特殊频率$\varOmega$ ,对应的本征值为1,即使在该外场为零的情况下也存在n1 ≠ 0的非平凡解。这些频率对应体系的激发能,相应的$n({r},\varOmega )$ 即为本征模。对于激发能和本征模的求解,1995年Casida[25]提出了著名的Casida方程
其中矩阵A、K的矩阵元为
式中:i和a分别为占据和未占据Kohn-Sham轨道的指标。而方程的解对应激发和退激发情形。若令K=0,则(14)式的本征值为Kohn-Sham激发能
${\omega _{ia\sigma }}$ 。根据Casida方程,只要满足如下条件就可以从TDDFT得到任意多体系统的严格激发能:(1)在知道基态DFT精确交换关联泛函的基础上,求出精确的Kohn-Sham基态以及所有占据、未占据的Kohn-Sham轨道及对应的本征值;(2)知道精确的交换关联核
${f_{{\rm{xc}}}}\left( {{r},{{r}^\prime },\omega } \right)$ ;(3)求解一个无穷维的赝本征值方程,由于矩阵元A、K依赖于$\varOmega $ ,该赝本征值方程需要迭代求解。在实际应用中,以上这些要求是不可能满足的,只能近似求解。一种近似求解方法是令非对角的矩阵元K为零,而保持矩阵元A不变,从而解耦激发项和退激发项,即所谓的Tamm-Dancoff近似(TDA)[26]。当激发频率不靠近零,对应分子、半导体及绝缘体情形时,TDA较为有效,有助于补偿交换关联泛函近似带来的误差。
TDDFT与线性响应理论的结合常称作线性响应含时密度泛函(Linear-Response Time-Dependent Density Functional Theory,LR-TDDFT),主要用于处理体系在微弱外场下的响应,此时体系偏离基态不远,用线性响应理论可以很好地描述,这正是许多谱学表征对应的情形。LR-TDDFT的另一个重要应用是计算体系的激发谱。理论上,这两方面性质可以由LR-TDDFT严格描述;在实际计算中,在合理的近似下LR-TDDFT对这些性质的计算可以达到较好的精度。虽然原则上实时求解含时Kohn-Sham方程组也可以得到包含线性响应在内的信息,但是一般情况下当体系偏离基态较小时,相比于显式地计算出随时间变化的电荷密度或波函数然后再计算它们相对于基态时的变化而言,直接计算这些相对基态较小的变化会更高效,计算也更容易。因此对于一般体系,LR-TDDFT是一种非常高效的方法,在大多数含时密度泛函软件包中已数值实现,应用非常广泛,以致过去提起TDDFT计算,一般指的就是LR-TDDFT计算。
不过LR-TDDFT也有缺点。从原理上讲,LR-TDDFT只适用于外界扰动不大的线性响应区域;当外场强度过大,用线性响应理论不足以描述时,不能再应用LR-TDDFT。随着激光技术的发展,许多情况都是LR-TDDFT无法描述的,例如短脉冲强场情形。另一方面,如果关心的正是体系的动力学性质,如去掉内层电子之后密度的振荡或原子电离过程等电子动力学情形、分子动力学这种原子核也在运动的情形,LR-TDDFT无法描述。除了原理上的困难,对于一些特殊的体系或性质,LR-TDDFT在数值计算上还存在一些困难,例如:对于大体系或电子态密度较高的体系,涉及的激发数目较多,当激发数目过高,如达到103~104时,对角化LR-TDDFT方程组的计算量为O(N6),LR-TDDFT很难应用;对于一些特殊性质,如计算X射线近吸收边结构时,其内层芯电子被激发,需要构造整个激发矩阵然后进行对角化,从而计算大量与问题无直接关系的根,且对角化所需计算量巨大,致使LR-TDDFT计算变得非常困难。
-
在LR-TDDFT不断发展的同时,实时含时密度泛函理论(Real-Time Time-Dependent Density Functional Theory,RT-TDDFT)也逐步发展起来并被广泛应用。相比于LR-TDDFT,RT-TDDFT的优势在于:从理论而言,RT-TDDFT可以无缝地处理从外场微扰到强场相互作用的情形,可以提取线性响应的相关信息(图1展示了RT-TDDFT与LR-TDDFT对比的一个例子[23]),并且当外场较大、非线性效应显著时,只能通过RT-TDDFT进行研究;RT-TDDFT还非常适合研究体系的动力学性质,包括电子与原子核耦合的动力学性质(例如用超短强激光脉冲研究化学键的断裂/形成、电荷转移等过程),RT-TDDFT对于研究实验探测背后的物理机制具有巨大的价值;从计算而言,对于大体系或电子态密度较高的体系,RT-TDDFT不存在LR-TDDFT所面临的困难,可以捕捉到较宽能谱范围内的电子激发,原则上体系的全吸收谱可以通过一次模拟获得(缺少对称性时需要3次模拟),包括从芯态到价态的激发。此外RT-TDDFT还有一个好处,不像LR-TDDFT那样处理交换关联泛函的泛函微分,这在应用某些复杂的交换关联泛函近似时非常有用。
RT-TDDFT也存在缺点。RT-TDDFT的计算量与所关心的问题和数值算法关系很大,大多数情况下只有模拟时长较长时,RT-TDDFT才能得到所需要的性质。
-
将TDDFT应用到实际问题时还需要一些必要的近似和高效的数值处理方式。实际应用TDDFT时主要考虑3方面问题:(1)为含时交换关联势寻找一个合理的近似,(2)对含时Kohn-Sham方程进行有效的数值求解,(3)从含时密度中得到人们感兴趣的物理量。对于这些问题,人们已经开展了大量的研究,使TDDFT得到了广泛的应用。
一般情况下,含时交换关联泛函与随时间变化的电荷密度和系统初始态相关。不过在某些常见情况下,交换关联泛函可以简化,例如初始态为基态的情形,这在许多实际问题中能够满足。此时根据Hohenberg-Kohn定理,初始态(即基态)完全由基态电荷密度(此时为初始电荷密度)决定,因此含时交换关联泛函完全由时变电荷密度决定,不用考虑初始态的影响。然而,即使在这种简单情况下,含时交换关联泛函依然非常复杂。除了存在与DFT中交换关联泛函一样的问题,其复杂性还源自其特有的时间上的非局域性:某一时刻t的交换关联泛函依赖于所有过去时刻(t′≤t)的电荷密度n(r′, t′)。为了实际应用,还需要作进一步近似。
对于含时交换关联泛函的近似,目前应用最广泛的是绝热近似(Adiabatic Approximation)。在该近似下,t时刻的交换关联泛函仅与该时刻的电荷密度相关,且泛函形式取为DFT中基态交换关联泛函形式,即
式中:
$v_{{\rm{xc}}}^{\rm{A}}$ 、$v_{{\rm{xc}}}^{{\rm{gs}}}$ 分别表示交换关联势的近似形式和基态交换关联势。按照该形式,只有当体系随时间变化非常缓慢且一直处于基态时,绝热近似才能很好地成立,然而一般情况下体系并不满足,因此含时交换关联泛函的绝热近似何时适用是TDDFT应用的一个重要问题。通过大量的实际应用,人们发现含时交换关联泛函的绝热近似在许多情况下都可以得到较好的结果。在很多情况下,在绝热近似条件下人们直接采用DFT中的局域密度近似(LDA),在空间上也将交换关联泛函近似为局域的,这就是所谓的绝热局域密度近似(ALDA)。当然也可以选用DFT中其他交换关联泛函近似,如广义梯度近似(GGA)等。
运用RT-TDDFT方法进行数值求解时,除了与DFT一样需要将Kohn-Sham轨道进行数值离散化,还需要将时间离散化,并在保证波函数模守恒的情况下求解含时Kohn-Sham方程,得到每个时间步上的Kohn-Sham轨道。
对于波函数随时间的演化,相邻时刻的波函数满足
式中:
${\text{δ}}t$ 为时间步长;$\hat U\left( {{t_n} + {\text{δ}}t,{t_n}} \right)$ 是将轨道从tn时刻传播到${t_n} + {\text{δ}}t$ 时刻的时间演化算符,计算时需要对其进行近似处理。当${\text{δ}}t$ 很小时,一种简单的近似为式中:
$ \hat H_{\rm s} $ 为体系哈密顿量。计算中体系哈密顿量有着自洽特性,即某时刻t的哈密顿量依赖于该时刻的电荷密度。因此数值计算中,在时间区间$\left[ {{t_n},{t_n} + {\text{δ}}t} \right]$ 内哈密顿量是未知的,只有在计算的时间步上哈密顿量是可以计算的。而某些处理时间演化算符的算法需要知道时间区间$\left[ {{t_n},{t_n} + {\text{δ}}t} \right]$ 内某一时刻的哈密顿量,这常常通过插值方法近似得到。虽然原则上为了充分的精确,该插值过程应该进行自洽计算,但是从实际计算效率考虑,往往直接采用二阶或二阶以上的插值,并且不进行自洽效验。对于时间演化,有多种方法进行数值近似处理[27],如经典的Crank-Nicolson算法、利用时间反演对称性的近似强制时间反演对称性(AETRS)算法等。当时间演化算符的近似处理涉及指数形式的哈密顿量时,还需要进行进一步的数值近似。最直接的方式是将指数函数进行泰勒展开,然后再进行截断,有些研究建议4阶为比较稳定的合理截断阶数。除此之外,还可以采用指数函数的Chebyshev展开,或者Lanczos方法。
有关波函数时间演化的数值方法正在蓬勃发展,有多种方法可供选择,不同的方法在达到同样精度时计算速度的差别可能很大,应用时需要根据体系特性对算法进行测试选取。
实际应用中人们关心的往往是可观测量的直接求解。虽然根据TDDFT,含时可观测量皆可由含时电荷密度决定,但是在实际计算中根据含时密度得到各种可观测量却存在难易之分,有些还需要作进一步近似。
有些可观测量能够严格地由电荷密度或电荷密度与Kohn-Sham轨道相结合得出。最直接的可观测量就是电荷密度本身;其他较易得到的物理量还有电荷的多级矩(如电偶极矩)、光吸收截面、原子体系的电离情况(通过电荷密度的空间积分得到),以及能够较直观衡量原子成键随时间演化性质的含时电子局域函数(TDELF,由含时电荷密度和Kohn-Sham轨道计算得出,如图2所示)[28]等。
还有些物理量则较难通过电荷密度得到,其中大部分与多体波函数或多体密度矩阵直接相关,包括光电子谱[29–31]、S矩阵[32]、转移密度矩阵[33]等。对于这些物理量,大多用含时Kohn-Sham轨道组成的Slater行列式代替真实体系多体波函数得到,但其可靠性没有严格的保证。
-
TDDFT经历几十年的发展,在很多领域取得了成功,然而所存在的不足限制了其应用。众所周知,当前被广泛应用的DFT并不完善,而TDDFT继承了DFT的优势和缺陷,在处理含时问题时也存在许多问题需要解决。目前,TDDFT正在蓬勃发展,从理论和应用两方面持续拓展,不断解决或改进发展过程中出现的问题。这部分内容非常庞大,本文仅针对其中一些感兴趣的重要进展进行简要介绍。
-
Runge-Gross定理奠定了TDDFT的基础,使其原则上可以处理时变标量场中多体系统随时间的演化,然而仍有许多重要的含时过程并不包括在内,例如含有变磁场或外部电磁场作用的情况。这些限制非常严重,因为严格地说TDDFT无法处理一般情况下电磁波与物质的相互作用。在实际应用中,通过一些近似方法变通地采用偶极近似处理某些特殊情况下电磁场与物质的相互作用,从而绕开该限制。对于一般情形,必须拓展新的理论以克服TDDFT的限制,从而处理矢量场形式的电磁场。为此,人们发展了含时电流密度泛函理论(TDCDFT)。
对于普适电磁场情形,通过以下简单的分析解释引入电流密度的必要性。多体系统在标量场v(r, t)和矢量场A(r, t)下的哈密顿量
$\hat H$ 的一般形式为式中:
$\hat W$ 为电子间的相互作用。由该哈密顿量出发可以求出含时多体波函数,进而得到电荷密度n(r, t)和电流密度j(r, t)随时间的变化。需要注意电流密度是一个矢量,总可以分解为纵向和横向分量。根据电荷密度的连续性方程,电流密度的纵向分量与电荷密度相关,而横向分量则与电荷密度无关,因此两个体系可以有相同的含时电荷密度、不同的横向电流密度。对于这样的两个体系,显然TDDFT无法描述。这是因为若TDDFT成立,则电荷密度相同,进而得出外势场相同,那么同一外势场对应两个不同的电流密度,出现v-representability问题[34]。为此,需要引入更多的自由度描述体系,此时势场为(v, A),为四分量。可以选取电荷密度n与电流密度j共同与之对应,即与TDDFT中势之间可以相差常量类似,在TDCDFT中可以通过规范变换相联系的电磁场被视为等效。
式中:
$\varLambda ({r},t)$ 为任意Well-Behaved的在初始时刻为零的规范函数。这一点在实际计算中非常有用,人们往往通过选取合适的规范函数来简化计算。对于TDCDFT,有类似的推广的van Leeuwen定理[35]作为其理论基础。同样,在TDCDFT框架下也有对应的含时Kohn-Sham方程
式中:有效标量势vs(r, t)与之前TDDFT一样,而有效矢量场As(r, t) = A(r, t) + Axc(r, t)。
TDCDFT是对TDDFT的一大推广,克服了TDDFT的一些固有缺陷,使得原则上可以处理普遍情况下的电磁波、含时磁场及矢量场。除此之外,它还有一些别的优点,例如对于一些情形,虽然原则上TDDFT也能够处理,但是如果用电流而非电荷密度作为基本参量,将会更好地构造包含动态效应的交换关联泛函[36–37]。
TDCDFT还将TDDFT的应用体系推广到一般情况的周期体系,对于实际应用具有很大的价值。在TDDFT的基石Runge-Gross定理的最初证明过程中,其关键步骤用到了在无穷远处面积分为零的边界条件,由此把体系的大小引入证明:对于有限体系,如原子分子体系,该条件是满足的;而对于无穷大体系,如拥有周期晶格的固体,该条件并不满足,故原始Runge-Gross定理的证明对固体不成立。后来人们将其进行推广,结果表明如果外势场具有周期性,那么Runge-Gross定理对固体依然成立[38]。然而周期体系的Runge-Gross定理存在很大的缺陷,即对于均匀外场时不成立,从而限制了其应用,因为该情况正对应了被广泛采用的长波近似。与之相对,TDCDFT则完全没有此限制[35]。
-
除了原子分子体系,金属和半导体等固体体系在电磁场中的响应也是人们非常关心的问题。在理论基础得以保证的情况下,对周期体系的含时密度泛函计算,特别是实时含时密度泛函计算也得到蓬勃发展。自2000年以来,Bertsch和Yabana等[39–41]、Rubio等[42–44]在该领域不断地推进,在相关理论、软件开发和应用方面取得了较大进展。
在实际计算中的许多情况下晶格常数远小于光波长,可用长波极限近似实际情况以简化计算,此时晶格中的电子相当于处于随时间变化但空间均匀的电场中。在一般原子分子等有限体系中,常常对此采用偶极近似(即长度规范)。然而如果将其应用在晶体中,对应的电势场为空间线性的,破坏了哈密顿量的空间周期性,如图3所示。为了解决该问题,人们利用之前所提的规范变换,通过选取合适的矢量势A(r, t),使标量势为零,保证哈密顿量的周期性,即所谓的速度规范。此时矢量势A可选为(设电场沿z方向,电场强度为E)
由此,只需要求解空间均匀的矢量场下的含时Bloch方程组即可。
需要注意:固体计算大多需要考虑宏观尺度下的物理量,特别是当外场非常强时,这是因为此时电位移和电场强度不再满足简单的线性关系,必须考虑麦克斯韦方程组与含时薛定谔方程的耦合。在该问题中有两个相差巨大的空间尺度,一个是微米量级的电磁场波长的宏观尺度,另一个则是埃量级的微观尺度。对于此类问题,Yabana等[41]在2012年提出了一种多尺度耦合的解决方法:先在宏观尺度上划分多个格点,在每个宏观尺度格点上考虑一个微观晶格,在晶格内应用TDDFT求解Kohn-Sham轨道,并得到相应的电流密度,通过积分和空间平均得到该宏观格点处的电流;然后在宏观格点上应用麦克斯韦方程组,求出宏观矢量场;最后根据每个格点的矢量场,得到每个微观晶格内的矢量场,从而实现信息交换和宏观-微观的耦合求解。该计算的计算量非常巨大,即使是低维模拟,也需要应用超级计算机的巨大计算资源。
-
与DFT一样,在TDDFT中交换关联泛函的近似从根本上决定了计算的准确程度。目前仍无法找到交换关联泛函的确切形式,只能近似地构造,这必然影响计算的准确性。在实际计算中,对于不同类型的体系,不同交换关联泛函形式有着不同的表现。TDDFT中的交换关联泛函与DFT一样也在迅速发展,一方面不断地吸取DFT中交换关联泛函发展的最新成果,另一方面根据实际计算所发现的问题在原有交换关联泛函的绝热近似基础上不断推进。近年来对许多重要典型问题的描述上,通过交换关联泛函的发展已经取得了不小的进步。
有一类很重要的激发:电荷从体系中的某一个区域移动到空间上分离的另一个区域,该过程在许多体系中都会发生,例如在复杂的分子复合体中或从分子的一个基团转移到另一基团。采用LR-TDDFT处理时发现,常用的交换关联泛函近似不能很好地描述该现象[45–47]。对于较简单的两部分空间相隔较远的体系,LR-TDDFT给出的电荷转移能(Charge-Transfer Energy)简化为施主和受主的Kohn-Sham轨道能级差;而DFT的常用近似(如LDA、GGA等)在处理电荷密度变化较大的情况时,轨道能量不准,从而大大低估电荷转移能,误差可达电子伏特量级。
为了更好地应用TDDFT处理电荷转移类问题,人们发展了多种方法,例如:Casida等[48]、Hu等[49–50]发展并应用了新的包括分数占据数的LR-TDDFT,改善了对电荷转移的描述。另一类方法是修正交换关联泛函的长程行为,使其具有正确的长程渐进行为,从而更好地描述能量较高的离域态性质。采用的方式为杂化密度泛函(Hybrid Functional),其长程部分用轨道交换作用形式表示,范围分离的杂化密度泛函(Range Separated Hybrid Functional)显著地提高了电荷转移的描述精度[51–55]。该泛函通过参数分离交换关联泛函的短程和长程部分,更好地通过轨道交换描述长程部分;但是参数的选取标准存在一些问题,大多依赖经验。近年来Baer等[56–57]提出参数不是普适的,而是依赖于体系的性质和结构。对于具体的某个体系,可以通过一些标准定出参数,例如所选参数应使体系的电离能等于最高占据态(HOMO)能级的相反数。通过这种依赖于系统的参数优化选取,进一步改善了电荷转移体系激发的描述,如表1[57]所示,其中B3LYP是一种常用的固定参数的杂化交换关联泛函,BNL是一种优化参数的范围分离杂化交换关联泛函,
$\gamma $ 为优化参数。除了电荷转移过程,还有其他一些性质(如Rydberg态等)也在采用范围分离的杂化交换关联泛函后计算精度得以大大提高。另外,除了原子分子体系,杂化交换关联泛函还对固体的含时密度泛函计算有所改善,例如:它能有效修正一般常用的交换关联泛函LDA和GGA等对半导体或绝缘体带隙的低估,提高光学带隙的计算精度,改进涉及带间跃迁的相关谱性质的计算。
除了应用DFT交换关联势的发展成果,TDDFT的交换关联势还有其自身的特点,其中重要一点是含时交换关联泛函的记忆性。原则上,它与过去所有时刻全空间的电荷密度相关,然而考虑到构造的复杂性,实际中大多采用所谓的绝热近似,仅考虑当前时刻的电荷密度对含时交换关联的影响。实践证明,绝热近似对于许多性质的计算精度是可以接受的。不过对于某些问题,绝热近似会带来较大的误差,此时必须考虑含时交换关联泛函的非绝热性。以下即为其中一些问题及其研究进展。
之前的理论研究表明,一个多体系统的激发谱可以由其响应函数
$\chi $ 的极点严格得到。原则上,通过TDDFT求解Casida方程就可以得到所有多体系统激发谱,然而应用交换关联核fxc的绝热近似得到的激发并不全面,有部分激发丢失。后来人们发现,激发谱的缺失与双激发(或多激发)特征有关系,也就是说多体体系的激发态包含显著的双激发组态或多激发组态成分。在TDDFT中,线性响应区域Kohn-Sham无相互作用体系不包含双激发或更高阶激发,因此Kohn-Sham响应函数${\chi _{\rm{s}}}$ 只在Kohn-Sham单激发处有极点,此极点比真实的多体响应函数$\chi $ 少。而应用Casida方程求解激发谱时,如果采用fxc的绝热近似,极点数目不会改变,仅仅发生平移,使其更接近真实多体系统的极点位置。为此,有必要引入非绝热、与频率相关的${f_{{\rm{xc}}}}(\omega )$ 带来更多的解,此时Casida方程变成非线性本征值方程,可以产生更多的极点。为了更好地描述分子的多激发性质,人们发展了一些非绝热的TDDFT,例如Dressed TDDFT方法[58]、基于多体理论的非绝热方法[59–62]。Dressed TDDFT方法最早于2004年由Maitra等[58]提出,此后不断发展,成为第一个成功依赖频率的交换关联核。为了克服绝热含时密度泛函近似无法很好处理多激发的缺点,Dressed TDDFT参考了多体波函数方法,将多体微扰理论与TDDFT相结合,其核心思想为:Dressed TDDFT的交换关联核可视为频率无关的绝热近似交换关联核与依赖频率的多体微扰核的杂化,其中多体微扰核可由二阶极化传播子方法得到。通过改进,Dressed TDDFT已经从最初的一次只能包含一个双粒子双空穴激发推广到多个双粒子双空穴激发。Dressed TDDFT被大量应用到需要处理多激发的领域,还与基于多体波函数的量子化学方法的基准计算进行了对比[63],取得了不错的结果。然而,在应用Dressed TDDFT时也出现一些问题,例如对于一些性质,本应该是单激发占主导地位,在应用Dressed TDDFT方法之后如果选取的交换关联泛函近似不恰当,会低估单激发的贡献而高估双激发的贡献。与量化基准计算的对比[63]表明,采用包含部分Hartree项贡献的杂化交换关联泛函与Dressed TDDFT联合使用可以得到较好的结果。
势能面是与体系多种基本性质相关的一个重要物理量。对于基态而言,基态势能面的全局最小点给出了体系的稳定几何构型,鞍点提供了过渡态的信息,并且基态势能面还包含原子间相互作用信息,可用于提取分子动力学所需的参数。激发态势能面也同样重要,在化学反应、光化学过程以及各类谱中发挥关键的作用[64–65]。基态势能面原则上可由DFT得到,而激发态势能面则可由TDDFT得到。
然而在实际计算过程中发现,即使采用具有正确长程行为的交换关联泛函,TDDFT计算得到的激发态势能面依然存在不小的误差,特别是当基态本身具有多组态特性时,两个势能面变得简并且相互靠近,形成所谓的圆锥交叉。对于这种拓扑结构的描述,TDDFT存在非常严重的问题[66–68]—在交叉点附近经常给出错误的拓扑结构,这与常用的绝热近似无法描述双激发有很大关系。频率相关的交换关联核
$f_{\rm{xc}}(\omega)$ 可以更好地解决该问题[69]。在周期体系中,半导体和绝缘体的光谱性质与交换关联核fxc的长程行为紧密相关,此时fxc在动量空间中的表示
${f_{\rm{x}}}\left( {q,G,{G^\prime },\omega } \right)$ 更便于应用。其中一类重要的极限情况是交换关联核$f_{\rm{xc}}\left(q, G, G^{\prime}, \omega\right)$ 在G = G′= 0(称为交换关联核的头)且q→0时的极限(该极限对应实空间中无限远的距离)。对于一般常用绝热近似下基于LDA或GGA的局域或半局域交换关联核,其交换关联核的头为一有限值,因此当q→0时,交换关联核的头对Casida方程中K项的贡献为零,说明Kohn-Sham谱的变化来源于交换关联核的其余部分。Gonze等[70–71]指出,为了正确地描述绝缘体的介电性质,当q→0时交换关联核的头应是q–2的发散行为。在这种情况下,交换关联核的头对Casida方程中K项的贡献相比于其他部分占决定性作用,而不是通常近似下的贡献为零。交换关联核的长波行为对原子分子这种有限体系的低能激发不太重要,但是对于扩展的周期体系,为了得到正确的谱,必须使交换关联核具有正确的长程行为[72–73]。从图4可以看到,交换关联核的头的贡献为零的ALDA不但错误地估计了光学带隙,而且没有很好地描述带隙附近的强激子峰。为了更好地描述绝缘体的光学性质,可以对交换关联核进行修正,使其头部有一有限值贡献。一种简便的方法是构造所谓长程修正(LRC)的交换关联核[71],如近期发展的Bootstrap核[74–75],它拥有正确的长程行为,并且计算表明在很大范围内的固体谱学计算中表现良好,如图5所示。
-
除了不断地改进理论和方法、提高计算精度,人们还将TDDFT应用到更广泛的领域。之前的研究大多在线性响应领域,此时外场较小,对系统的作用可视为微扰,应用TDDFT可研究弱场下的响应和体系的本征激发谱。然而,超短超强激光脉冲(其外场作用强度可以与材料内部的库仑作用相比拟)以及一些极端情况(如某些极端条件下的热效应甚至可以与库仑作用相比拟),对理论计算提出了新的要求。原则上TDDFT可以处理非相对论情况下任意大小的外场与物质的相互作用,因此近年来不断发展,并在极端环境领域开始应用。
引起人们关注的强场下高次谐波的产生是一个典型的无法用微扰描述的物理问题,有着巨大的基础研究和应用价值。之前人们已对原子分子等有限体系中的高次谐波的产生做了大量的理论和实验研究。最近Tancogne-Dejean等[76]应用TDDFT对块材硅中高次谐波的产生进行了深入研究,结合解析模型和第一性原理计算,给出了周期性固体体系相对于原子分子体系在高次谐波产生上的特点,并将其与能带结构联系起来(见图6)。在计算方面,综合了DFT和TDDFT的结果,将第一性计算扩展到强场与无限体系相互作用的领域。
除了强场领域,近年来TDDFT还被应用到极端条件下的物态研究,如2016年实时有限温度TDDFT被首次应用于温稠密物质[77]。温稠密物质包含的范围很广,一般被视为凝聚态与理想等离子体的中间态,其中费米简并依然存在,且热效应和库仑作用的量级相同。X射线汤姆逊散射谱是探索温稠密物质的重要工具。通过对比实验测得的X射线汤姆逊散射谱线和理论计算,可以得到温稠密物质的温度、密度、电离度等各类物性参数,因此理论建模的准确性非常重要。在大多数传统模型中,Chihara分解被广泛采用,其中电子被划分为紧绑定、松绑定和自由电子。对于温度和压力较高的温稠密物质,该分解的可信度存疑。对此,Baczewski等[77]采用有限温度TDDFT结合投影缀加平面波方法研究温稠密态的铍的动力学性质。计算中没有芯电子被冻结,因此不再区分束缚态电子和自由电子;Mermin方法被用于处理初始态及之后轨道的演化。研究显示:相对于传统的唯象理论模型,第一性原理TDDFT计算具有独特的优势,尤其对于束缚电子和自由电子划分较模糊的体系。
近年来中国工程物理研究院也应用TDDFT对温稠密物质进行了研究。张平等[78]应用TDDFT的微扰形式研究了温稠密态铝的X射线汤姆逊散射谱(见图7),并将其应用于电子温度的测量,其中温度采用Mermin有效温度形式,电子在X射线电场下的散射通过含时密度泛函线性微扰响应方式研究。通过计算给出电子温度上限为2000 K左右,远小于通过细致平衡关系估算得到的60 000 K,表明X射线自由电子激光被用于结构测量时对样品的加热效应可能小于预期。
-
除了对TDDFT自身的研究,人们还将TDDFT与其他学科相结合,开辟了一些新的发展方向。近年来,统计学习和大数据迅速兴起,渗透到日常生活和科研生产的各个领域。在材料模拟领域,统计学习与已有方法相结合,成为平衡计算精度和计算复杂度的一种新型计算手段。相对于量子化学的基于波函数的组态相互作用、耦合团簇法等,TDDFT具有计算量较小的特点,是计算大体系可行的第一性原理解决方案。如前所述,现有的计算量较小的含时密度泛函绝热近似不能很好地描述电荷转移及具有双激发特征的性质,使得TDDFT在光化学等领域的应用受到制约;通过与大样本分子数据库中精度较高的量化方法的计算结果进行对比开展统计学习,有望系统地修正TDDFT的计算结果,使其对数据库之外的体系也具有一定的预测性。2015年Ramakrishnan等[80]比较了20 000个有机小分子的TDDFT与较高精度的二阶耦合团簇方法在计算较低的单态-单态垂直电子谱等激发态性质的结果,并对计算结果的差异进行了统计学习,发现统计学习预测结果的误差随着训练集的增大而单调降低,对于有10 000个分子的训练集,预测激发能的误差可小于0.1 eV(考虑到TDDFT自身的系统误差带来的偏移,结果可以进一步修正以提高精度)。该结果表明对于各种类型的误差来源,如含时密度泛函绝热近似的双激发系统误差、基组不完备的数值误差、体系的电荷转移特性引起的误差等,统计学习手段都可以加以修正。现在这方面研究方兴未艾,随着数据库的不断增大,数据驱动的以TDDFT计算为基础的激发态性质建模也逐渐成为一种重要的新兴研究手段。
-
TDDFT已为多种情形下的理论计算奠定了坚实的基础,通过适当的近似使数值计算成为可能,计算结果与实验结果在多个方面都符合得很好;然而,随着应用的不断深入,人们发现了不少现阶段仍无法很好处理的难题,对相关的理论和计算方法提出了新的挑战。
目前TDDFT在实际应用时广泛采用绝热近似,该近似中含时交换关联泛函没有考虑过去时刻的记忆效应,而是采用基态DFT中的交换关联泛函,因而DFT中交换关联泛函的精确程度对TDDFT计算精度的影响很大。DFT中的交换关联泛函仍不完善,在处理一些体系以及描述某些性质时误差较大,例如密度泛函的局域密度近似和广义梯度近似在计算半导体或绝缘体的带隙时会严重低估带隙,甚至导致定性的计算错误。为此,人们提出了各种改进方案,如杂化交换关联泛函、meta-GGA,然而这两种方法的常用方案往往需要选取经验参数,不利于方法的可迁移性和预测性。DFT在这方面的误差与常用的交换关联泛函不能很好描述化学势的不连续性以及不正确的长程渐近行为有关。除了带隙,DFT也无法正确描述电荷迁移、Rydberg态等性质。这些都直接影响采用相同交换关联泛函形式的TDDFT。因此从某些角度讲,TDDFT和DFT在交换关联泛函方面具有不少相同的挑战。
其中一项挑战是目前DFT和TDDFT处理起来都非常困难的体系—强关联体系。在强关联体系中,由于电子之间存在很强的关联效应,因此出现许多新奇的特性,引起了基础研究、国防工业领域的极大关注。然而电子强关联效应却为理论和计算处理带来了巨大的困难。DFT计算中常用的局域密度近似和广义梯度近似是平均场方法,不适合处理强关联体系。后来发展的较为简单的DFT+Hubbard U的修正效果也不理想。近年来DFT与Gutzwiller方法以及动力学平均场方法相结合,对强关联体系的计算取得了较大的突破,但却依然没有达到理想的精度,且后者的计算量非常巨大。DFT在这方面依然需要寻求突破,至今仍没有合适的交换关联泛函或方法来处理该类体系。TDDFT的任务则更加艰巨,没有直接可以从DFT借用的交换关联泛函和方法,在研究强关联体系的一些特定性质时,还需要超越DFT,这也为它带来了更高的挑战。
从TDDFT本身来看,一般情况下其含时交换关联泛函与初始状态及所有过去时刻的电荷密度有关,然而目前广泛采用的绝热近似却忽略了这一点,虽然能够较好地处理以单激发为主的系统,但是一旦系统出现多激发,则描述效果较差。发展超越绝热近似的含时交换关联泛函是目前解决相关问题所面临的一个挑战。对此,人们已经取得了一些进展,如Dressed TDDFT[58],然而依然需要改进,尚未成为应用的主流。此外,只有当初态为系统基态时,含时交换关联泛函才严格地只依赖于电荷密度;当初始状态非基态时,原则上还应考虑含时交换关联泛函对初始状态的依赖关系,在这方面人们考虑得较少。
事实上Runge-Gross定理的初始状态依赖性可能促进新的交换关联泛函的发展。对于真实的有相互作用的体系,给定一个初始态,则有无穷多个可能的Kohn-Sham初始态具有相同的初始电荷密度。如果能够针对所研究的性质选取恰当的Kohn-Sham初始态,即使依然采用含时密度泛函的绝热近似,也可能显著改进计算结果,在这方面已有一些初步研究[81–83]。就算在线性响应区域,选取激发态作为Kohn-Sham初始态也可能对微扰导致的激发频率产生较好的改善。例如,选取一个单激发态为Kohn-Sham初始态,则在单激发态基础上的单激发就可以形成一次双激发,从而避免显式地引入非绝热交换关联泛函。
基态的DFT已经拓展到有限温度情形和具有相对论效应的体系,然而目前相应的TDDFT发展相对缓慢。相对论性的TDDFT的理论基础还有待严格的证明。这些拓展对于将TDDFT应用到极端条件具有非常重要的意义,例如:有限温度的TDDFT可用于研究非平衡态热力学和含时热力学系综,有助于计算热输运等性质;相对论性的TDDFT可用于研究重核材料以及量子电动力学。
分子动力学是研究材料微观性质的重要计算手段,而第一性原理分子动力学因不借助于经验势和预测性更具优越性。基于DFT的第一性原理分子动力学采用Born-Oppenheimer近似,从基态势能面得到力的信息。然而在诸如超快激光驱动的分子解离等情况下,Born-Oppenheimer近似不再是一个很好的近似,此时需要更准确地考虑电子与原子核之间的耦合。原则上TDDFT能够给出激发态的势能面,因此它可以与分子动力学相结合,研究非绝热动力学性质。目前在该领域已经有多种不同的结合方案,如传统的Ehrenfest近似[84–87]、Surface-Hopping方法[88–90]等。然而,这些方法都有其不足之处,如都没有考虑核本身的量子效应,导致无法描述核之间的相干等信息。在这方面,TDDFT具有巨大的应用前景,但依然需要新的理论和算法来完善基于TDDFT的非绝热分子动力学。
采用第一性原理计算时,通常希望能够计算的体系越大越好。基于波函数的量子化学方法受其理论基础的限制,无法实现大体系计算。在这方面,基于电荷密度的DFT和TDDFT是目前大体系计算最可行的方案,人们也为此做了大量研究。得益于线性标度方法,DFT已经可以用于上万个原子甚至更大体系的第一性原理计算。而TDDFT依然需要进一步发展,特别是一些特殊情况,例如:当大体系的非微扰效应显著时,需要RT-TDDFT计算;必须考虑光传播效应时,若采用与麦克斯韦方程组相耦合的方式,则由于两套理论不同的空间尺度,计算量非常巨大。因此,发展高效的算法或多尺度方法同样是TDDFT应用的一个挑战。
根据存在于多类体系的近视(Nearsightedness)特性,处理大体系时一种常用的做法是分而治之:将体系划分为许多小体系,然后通过计算小体系的性质,并考虑它们之间的耦合,得到大体系的性质。这种策略已被应用于大体系基态性质的计算中。应用TDDFT计算大体系激发态时,也可以采用类似做法,在LR-TDDFT和RT-TDDFT方面人们已经开展了相关工作[91–93]。这种划分子系统的方法除了具有计算优势外,还能够从物理上深入理解子系统之间的耦合以及子系统和完整系统性质之间的联系。不过这种策略所能处理的体系类型有一定限制,如何进行拓展或提高精度需要进一步探索。
除了大体系的计算,在应用方面开放体系计算也是一个重要问题。对于原子分子这类有限体系或者周期性固体体系,目前都有较成熟的处理方法,然而仍存在多类开放体系,其中一类常见的重要情形是在纳器件领域,分子两端连在电极上,电极可视为电子库的半无限体系。这类体系的计算不同于分子或固体体系,需要建立新的理论框架。DFT在该领域已经有一套框架,例如采用密度泛函结合非平衡格林函数研究稳态的输运性质。TDDFT可用于研究开放体系的动态性质,目前也提出了不同的研究方案,然而其可靠性仍然存在争议,需要进一步发展。
-
与DFT一样,TDDFT作为一种高效的第一性原理计算方法,从诞生之初就备受关注,经过长期持续发展,TDDFT已成功应用到诸多领域。由于其巨大的应用前景,未来TDDFT仍将继续蓬勃发展。
在理论基础方面,有望进一步将Runge-Gross定理和van Leeuwen定理推广到更一般的情形,为研究更一般情况下多体体系电子动态性质提供坚实的理论基础。同时,TDDFT也将被推广到更多的场景,如有限温度甚至温稠密状态、相对论效应显著的情况等,这些都需要理论的进一步拓展。
在应用方面,一个非常重要的方向是不断提高计算精度,同DFT的目标一样,希望最终实现化学精度的计算。为此发展更优异的含时交换关联泛函至关重要,如何更好地考虑双激发或更高激发是一个重要的努力方向。对于一些复杂体系(如强关联电子体系),超越简单理论模型实现强关联材料的高精度含时密度泛函计算也是一个重要的发展方向。应用方面的另一个重要方向则是在保证一定精度的情况下不断提升所能处理的体系大小,无论研究材料的微介观性质,还是生物大分子体系,抑或光与物质相互作用的宏观效应等,都具有重要的实际意义。为此,在提升计算机硬件性能的同时,对处理方式(如多尺度耦合)以及数值算法提出了更高的要求。
随着TDDFT的不断发展,作为一种普适的第一性计算方法,TDDFT有望成为多个领域研究微观动态性质的一把利器,更好地解释和预言材料性质,并与不同尺度的方法相结合发挥更大的作用。
含时密度泛函理论及应用的最新发展
New Developments of Time-Dependent Density Functional Theory and Its Applications
-
摘要: 密度泛函理论在材料计算研究领域得到了广泛的应用,然而它无法处理含时问题和材料的激发态性质。Runge-Gross定理奠定了含时密度泛函理论的基础,为研究这两类问题提供了有效的手段。经过三十多年的发展,含时密度泛函已被应用到量子化学、材料计算等多个领域,人们也更加了解其优势和不足。目前,含时密度泛函理论和方法仍在迅速发展。本文简要回顾含时密度泛函方法的发展历史,介绍近年来含时密度泛函在理论和应用方面的一些重要进展,总结当前在含时密度泛函领域存在的重要难题以及面临的挑战,展望其发展方向和趋势。Abstract: Nowadays density functional theory which was introduced in the mid-1960s has wide applications in material simulations. However, it is not able to deal with time-dependent problems and excited properties of materials. Time-dependent density functional theory (TDDFT) based on Runge-Gross theorem, provides a viable way to deal with these two problems. After thirty years’ development, TDDFT has been widely applied to many fields, such as quantum chemistry and material simulation, and our understanding of its advantages and weaknesses also grows. To date, TDDFT theory and method still develop rapidly. Here a brief historical review of TDDFT is first introduced. Then it is followed by a discussion of recent important developments on theory and applications of TDDFT. Finally we summarize some important problems and challenges that TDDFT are facing and attempt to offer some thoughts about where TDDFT will be progressing.
-

-
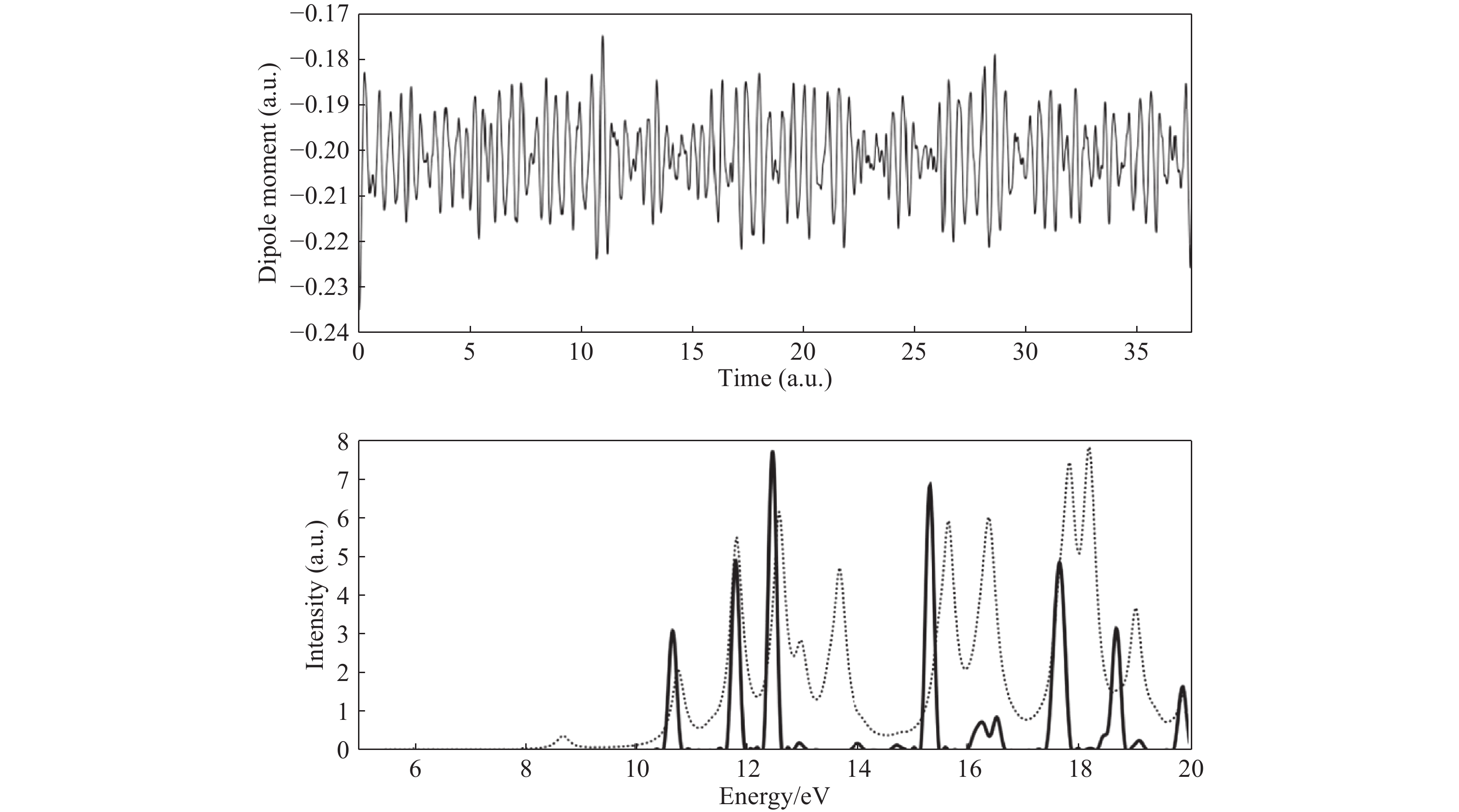
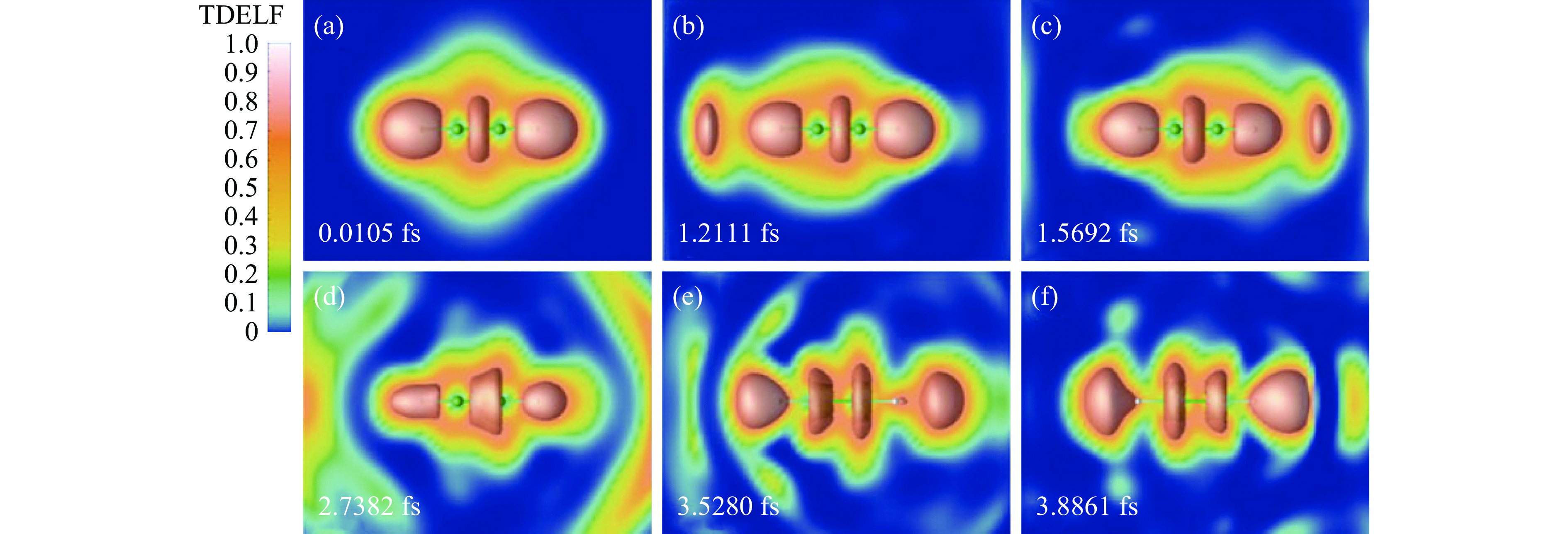
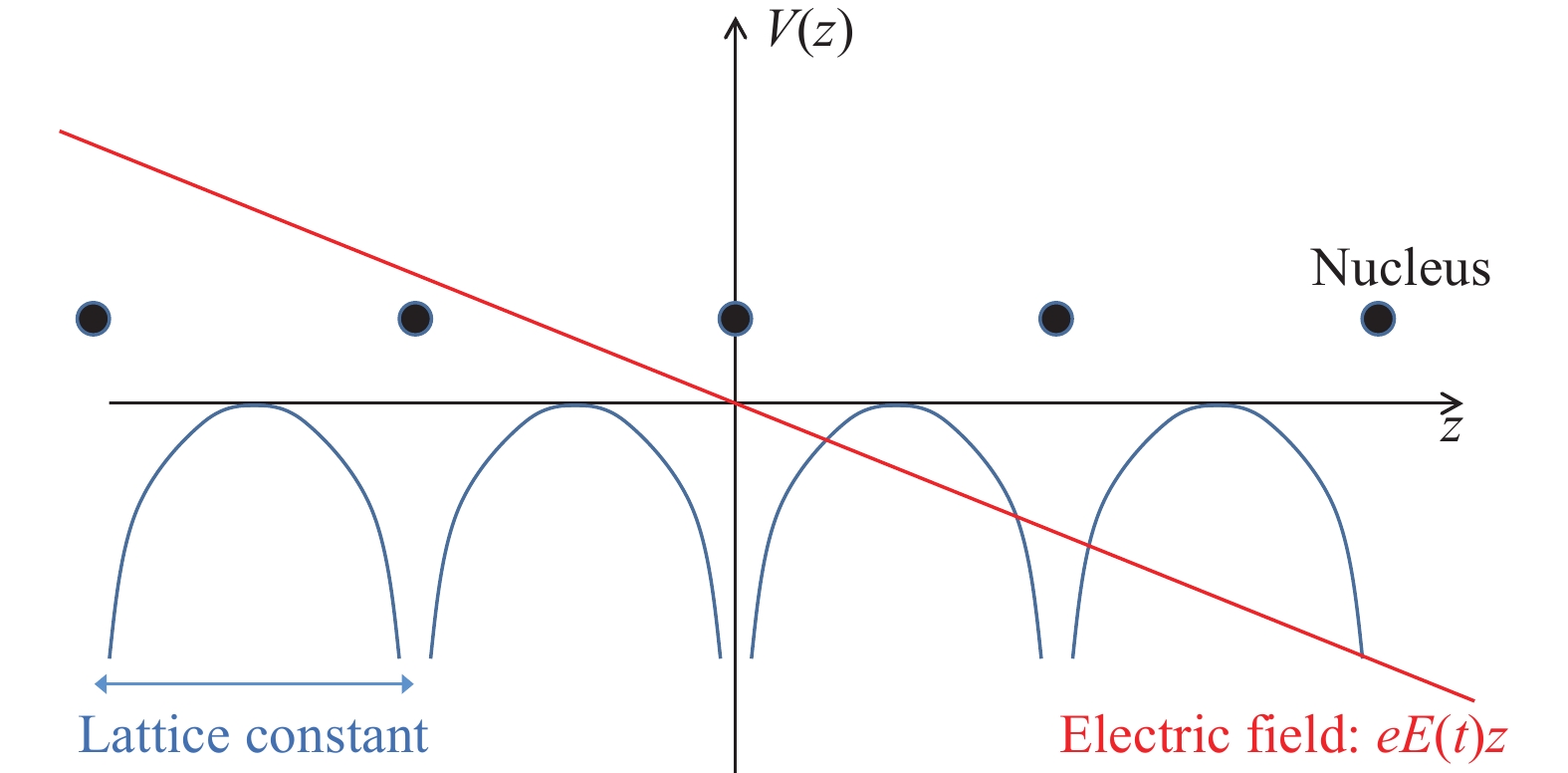
图 3 偶极近似破坏了晶体哈密顿量的空间周期性
Figure 3. Spatial periodicity of the Hamiltonian destroyed by the dipole approximation
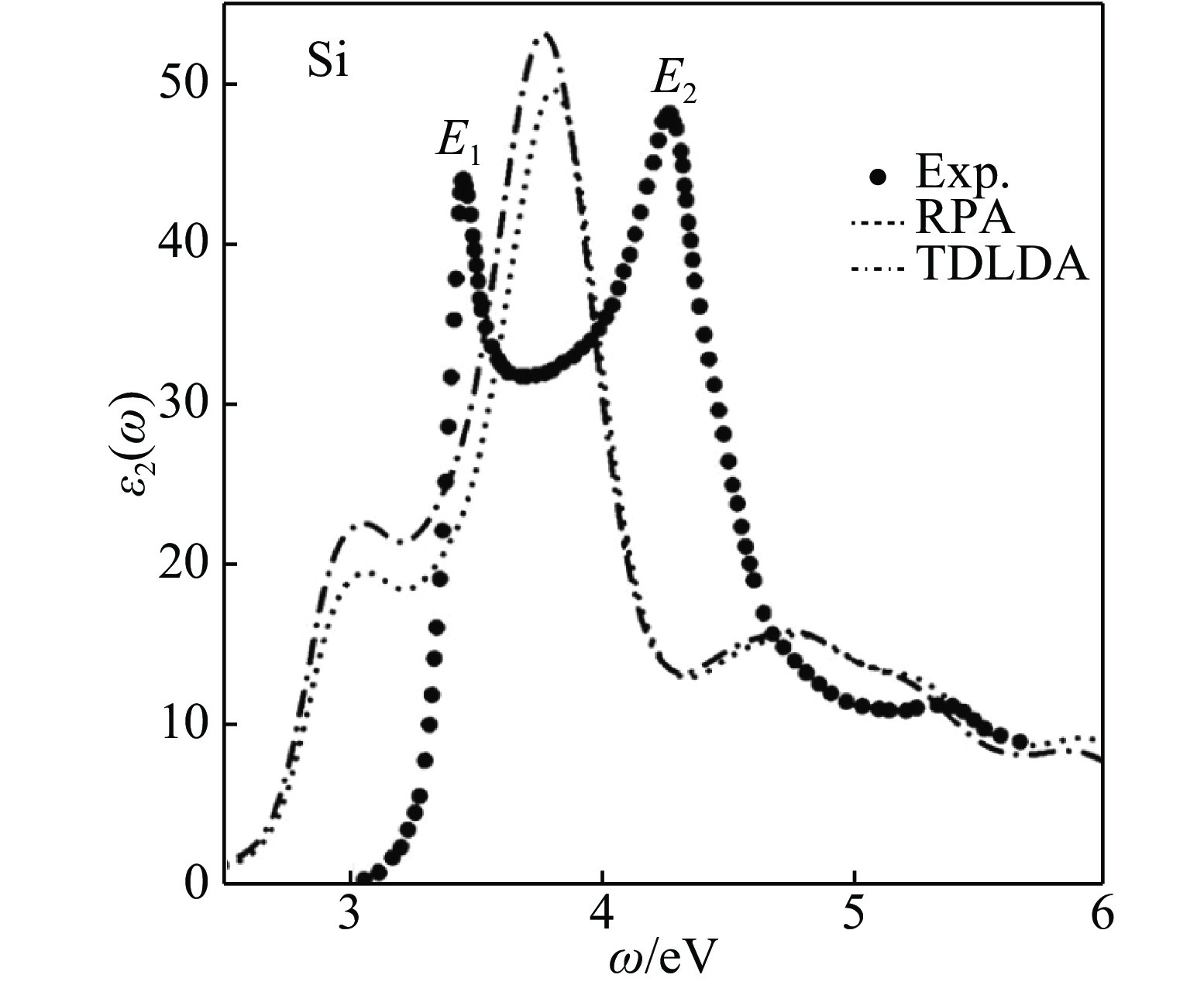
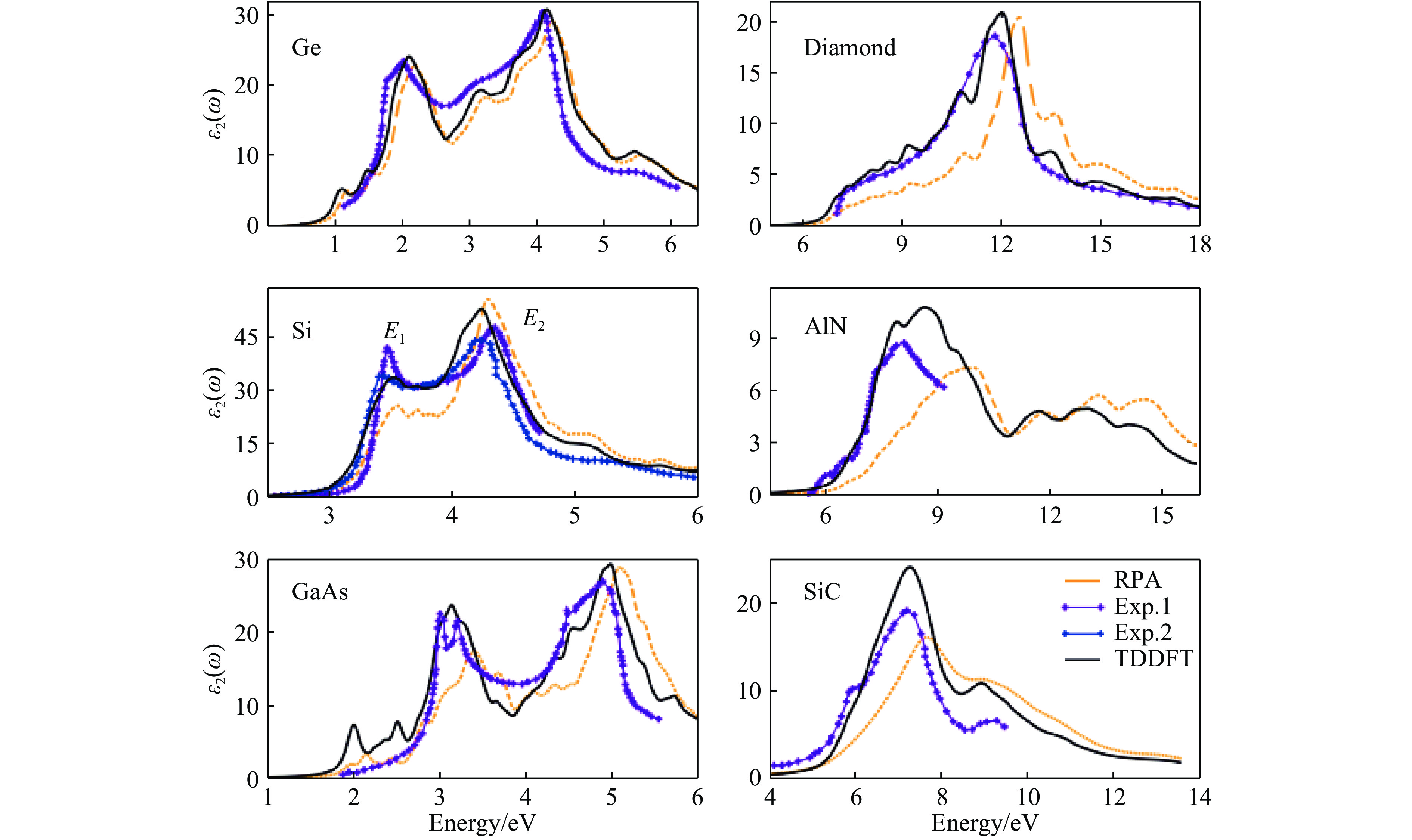
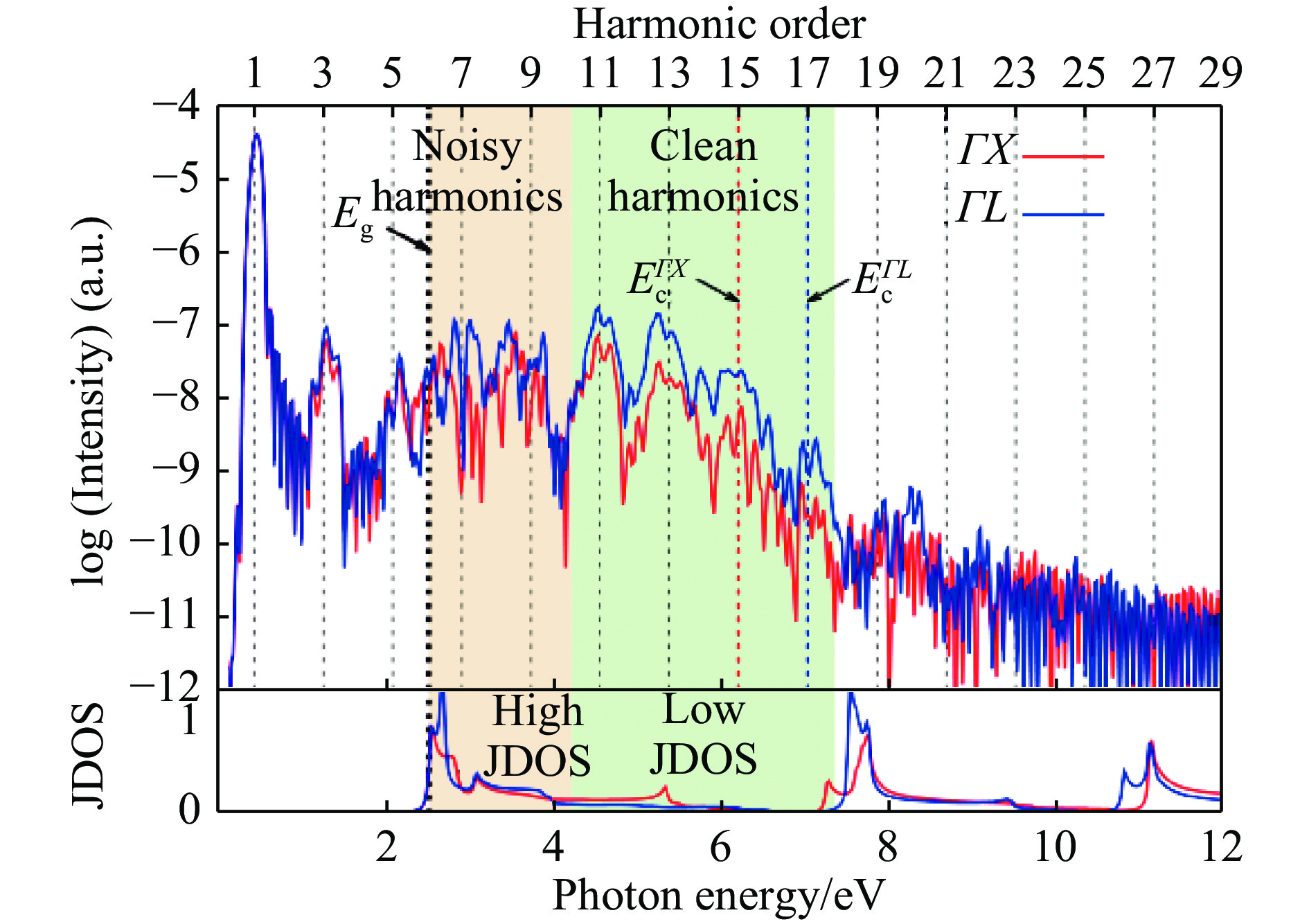
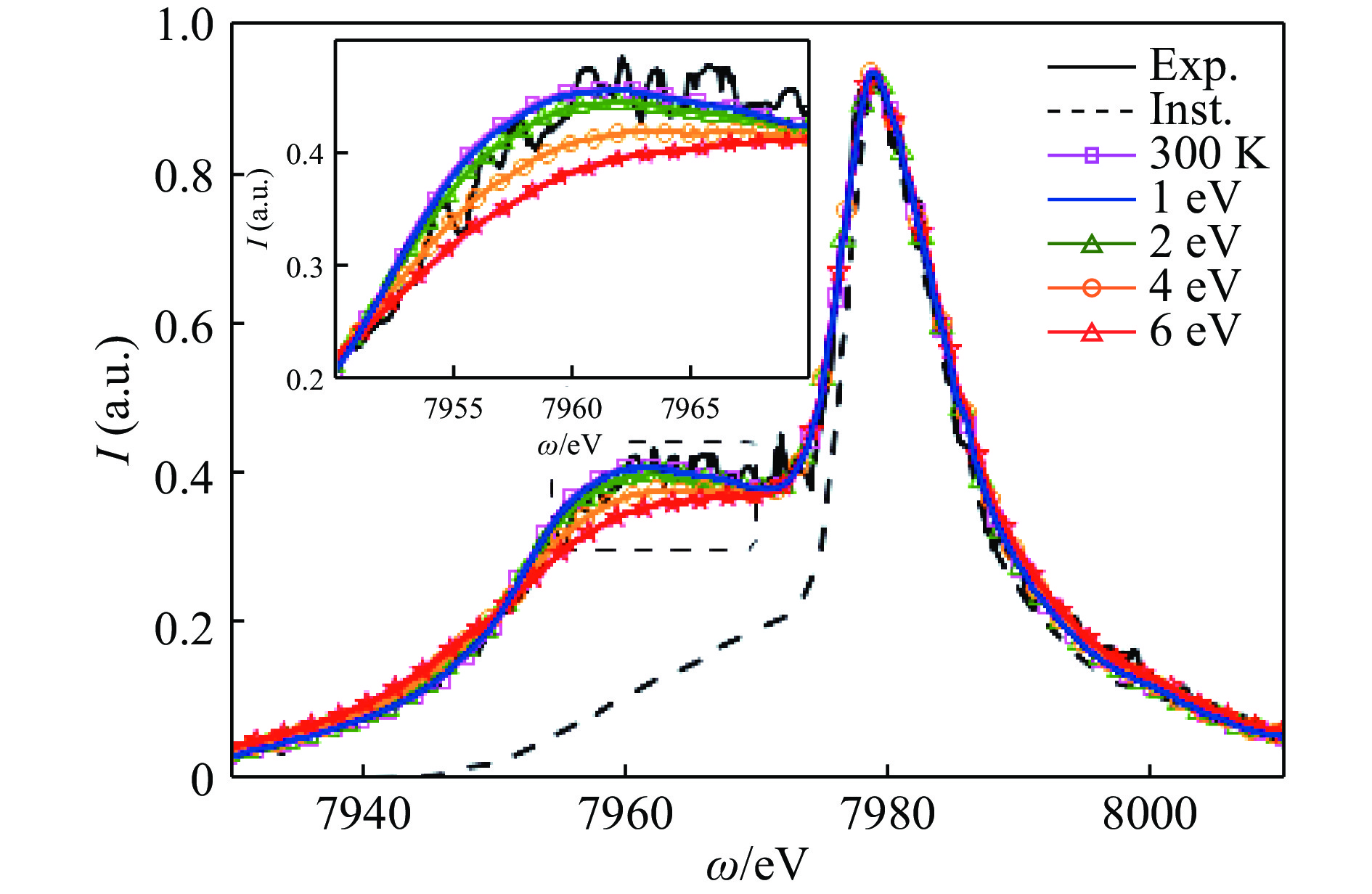
表 1 一些气相Ar-TCNE体系的激发能E(单位eV)和振动强度f的计算和实验对比[57]
Table 1. Excitation energies E (eV) and oscillator strengths f of several gas phase Ar-TCNE systems: theory and experiment[57]
Ar B3LYP BNL ( $\gamma $ =0.5)

BNL $\gamma^* $ 

Exp.[27] E f E $\gamma^* $ 

E f E f Benzene 2.1 0.03 4.4 0.33 3.8 0.03 3.59 0.02 Toluene 1.8 0.04 4.0 0.32 3.4 0.03 3.36 0.03 O-xylene 1.5 0 3.7 0.31 3.0 0.01 3.15 0.05 Naphthalene 0.9 0 3.3 0.32 2.7 0 2.60 0.01  下载: 导出CSV
下载: 导出CSV
-
[1] DIRAC P A M. Quantum mechanics of many-electron systems [J]. Proceedings of the Royal Society of London Series A, 1929, 123(792): 714–733. doi: 10.1098/rspa.1929.0094 [2] HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Physical Review, 1964, 136(3B): B864–B871. doi: 10.1103/PhysRev.136.B864 [3] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects [J]. Physical Review, 1965, 140(4A): A1133–A1138. doi: 10.1103/PhysRev.140.A1133 [4] BLOCH F. Bremsvermögen von Atomen mit mehreren Elektronen [J]. Zeitschrift für Physik A Hadrons and Nuclei, 1933, 81(5): 363–376. [5] PEUCKERT V. A new approximation method for electron systems [J]. Journal of Physics C: Solid State Physics, 1978, 11(24): 4945–4956. doi: 10.1088/0022-3719/11/24/023 [6] ZANGWILL A, SOVEN P. Density-functional approach to local-field effects in finite systems: photoabsorption in the rare gases [J]. Physical Review A, 1980, 21(5): 1561. doi: 10.1103/PhysRevA.21.1561 [7] DEB B M, GHOSH S K. Schrödinger fluid dynamics of many-electron systems in a time-dependent density-functional framework [J]. The Journal of Chemical Physics, 1982, 77(1): 342–348. doi: 10.1063/1.443611 [8] GHOSH S K, DEB B M. Dynamic polarizability of many-electron systems within a time-dependent density-functional theory [J]. Chemical Physics, 1982, 71(2): 295–306. doi: 10.1016/0301-0104(82)87030-4 [9] GHOSH S K, DEB B M. A density-functional calculation of dynamic dipole polarizabilities of noble gas atoms [J]. Theoretica Chimica Acta, 1983, 62(3): 209–217. doi: 10.1007/BF00548835 [10] GHOSH S K, DEB B M. A simple density-functional calculation of frequency-dependent multipole polarizabilities of noble gas atoms [J]. Journal of Molecular Structure: THEOCHEM, 1983, 103: 163–176. doi: 10.1016/0166-1280(83)85017-9 [11] BARTOLOTTI L J. Time-dependent extension of the Hohenberg-Kohn-Levy energy-density functional [J]. Physical Review A, 1981, 24(4): 1661–1667. doi: 10.1103/PhysRevA.24.1661 [12] BARTOLOTTI L J. Time-dependent Kohn-Sham density-functional theory [J]. Physical Review A, 1982, 26(4): 2243–2244. doi: 10.1103/PhysRevA.26.2243 [13] BARTOLOTTI L J. Variation-perturbation theory within a time-dependent Kohn-Sham formalism: an application to the determination of multipole polarizabilities, spectral sums, and dispersion coefficients [J]. The Journal of Chemical Physics, 1984, 80(11): 5687–5695. doi: 10.1063/1.446637 [14] BARTOLOTTI L J. Velocity form of the Kohn-Sham frequency-dependent polarizability equations [J]. Physical Review A, 1987, 36(9): 4492–4493. doi: 10.1103/PhysRevA.36.4492 [15] RUNGE E, GROSS E K U. Density-functional theory for time-dependent systems [J]. Physical Review Letters, 1984, 52(12): 997–1000. doi: 10.1103/PhysRevLett.52.997 [16] LEVY M. Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem [J]. Proceedings of the National Academy of Sciences, 1979, 76(12): 6062–6065. doi: 10.1073/pnas.76.12.6062 [17] LIEB E H. Density functionals for Coulomb systems [J]. International Journal of Quantum Chemistry, 1983, 24: 243–277. doi: 10.1002/(ISSN)1097-461X [18] VAN LEEUWEN R. Mapping from densities to potentials in time-dependent density-functional theory [J]. Physical Review Letters, 1999, 82(19): 3863–3866. doi: 10.1103/PhysRevLett.82.3863 [19] RUGGENTHALER M, GIESBERTZ K J H, PENZ M, et al. Density-potential mappings in quantum dynamics [J]. Physical Review A, 2012, 85(5): 052504. doi: 10.1103/PhysRevA.85.052504 [20] RUGGENTHALER M, VAN LEEUWEN R. Global fixed-point proof of time-dependent density-functional theory [J]. EPL (Europhysics Letters), 2011, 95(1): 13001. doi: 10.1209/0295-5075/95/13001 [21] ULLRICH C A. Time-dependent density-functional theory: concepts and applications [M]. OUP Oxford, 2011. [22] GIULIANI G, VIGNALE G. Quantum theory of the electron liquid [M]. Cambridge: Cambridge University Press, 2005. [23] ULLRICH C A, YANG Z. A brief compendium of time-dependent density functional theory [J]. Brazilian Journal of Physics, 2014, 44(1): 154–188. doi: 10.1007/s13538-013-0141-2 [24] GROSS E K U, KOHN W. Local density-functional theory of frequency-dependent linear response [J]. Physical Review Letters, 1985, 55(26): 2850–2852. doi: 10.1103/PhysRevLett.55.2850 [25] CASIDA M E. Time-dependent density functional response theory for molecules [M]//Recent Advances in Computational Chemistry. World Scientific, 1995: 155–192. [26] HIRATA S, HEAD-GORDON M. Time-dependent density functional theory within the Tamm-Dancoff approximation [J]. Chemical Physics Letters, 1999, 314(3/4): 291–299. [27] CASTRO A, MARQUES M A L, RUBIO A. Propagators for the time-dependent Kohn-Sham equations [J]. The Journal of Chemical Physics, 2004, 121(8): 3425–3433. doi: 10.1063/1.1774980 [28] BURNUS T, MARQUES M A L, GROSS E K U. Time-dependent electron localization function [J]. Physical Review A, 2005, 71(1): 010501. doi: 10.1103/PhysRevA.71.010501 [29] POHL A, REINHARD P G, SURAUD E. Towards single-particle spectroscopy of small metal clusters [J]. Physical Review Letters, 2000, 84(22): 5090–5093. doi: 10.1103/PhysRevLett.84.5090 [30] VÉNIARD V, TAIEB R, MAQUET A. Photoionization of atoms using time-dependent density functional theory [J]. Laser Physics, 2003, 13(4): 465–474. [31] DE GIOVANNINI U, VARSANO D, MARQUES M A L, et al. Ab initio angle-and energy-resolved photoelectron spectroscopy with time-dependent density-functional theory [J]. Physical Review A, 2012, 85(6): 062515. doi: 10.1103/PhysRevA.85.062515 [32] ROHRINGER N, PETER S, BURGDÖRFER J. Calculating state-to-state transition probabilities within time-dependent density-functional theory [J]. Physical Review A, 2006, 74(4): 042512. doi: 10.1103/PhysRevA.74.042512 [33] LI Y, ULLRICH C A. Time-dependent transition density matrix [J]. Chemical Physics, 2011, 391(1): 157–163. doi: 10.1016/j.chemphys.2011.02.001 [34] D’AGOSTA R, VIGNALE G. Non-V-representability of currents in time-dependent many-particle systems [J]. Physical Review B, 2005, 71(24): 245103. doi: 10.1103/PhysRevB.71.245103 [35] VIGNALE G. Mapping from current densities to vector potentials in time-dependent current density functional theory [J]. Physical Review B, 2004, 70(20): 201102. doi: 10.1103/PhysRevB.70.201102 [36] VIGNALE G, KOHN W. Current-dependent exchange-correlation potential for dynamical linear response theory [J]. Physical Review Letters, 1996, 77(10): 2037–2040. doi: 10.1103/PhysRevLett.77.2037 [37] VIGNALE G, ULLRICH C A, CONTI S. Time-dependent density functional theory beyond the adiabatic local density approximation [J]. Physical Review Letters, 1997, 79(24): 4878–4881. doi: 10.1103/PhysRevLett.79.4878 [38] MAITRA N T, SOUZA I, BURKE K. Current-density functional theory of the response of solids [J]. Physical Review B, 2003, 68(4): 045109. doi: 10.1103/PhysRevB.68.045109 [39] YABANA K, BERTSCH G F. Time-dependent local-density approximation in real time [J]. Physical Review B, 1996, 54(7): 4484–4487. [40] BERTSCH G F, IWATA J I, RUBIO A, et al. Real-space, real-time method for the dielectric function [J]. Physical Review B, 2000, 62(12): 7998–8002. [41] YABANA K, SUGIYAMA T, SHINOHARA Y, et al. Time-dependent density functional theory for strong electromagnetic fields in crystalline solids [J]. Physical Review B, 2012, 85(4): 045134. [42] MARQUES M A L, CASTRO A, BERTSCH G F, et al. Octopus: a first-principles tool for excited electron–ion dynamics [J]. Computer Physics Communications, 2003, 151(1): 60–78. [43] CASTRO A, APPEL H, OLIVEIRA M, et al. Octopus: a tool for the application of time-dependent density functional theory [J]. Physica Status Solidi (B), 2006, 243(11): 2465–2488. [44] ANDRADE X, STRUBBE D, DE GIOVANNINI U, et al. Real-space grids and the Octopus code as tools for the development of new simulation approaches for electronic systems [J]. Physical Chemistry Chemical Physics, 2015, 17(47): 31371–31396. [45] DREUW A, HEAD-GORDON M. Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin-bacteriochlorin and bacteriochlorophyll-spheroidene complexes [J]. Journal of the American Chemical Society, 2004, 126(12): 4007–4016. doi: 10.1021/ja039556n [46] HIERINGER W, GÖRLING A. Failure of time-dependent density functional methods for excitations in spatially separated systems [J]. Chemical Physics Letters, 2006, 419(4/5/6): 557–562. [47] AUTSCHBACH J. Charge-transfer excitations and time-dependent density functional theory: problems and some proposed solutions [J]. ChemPhysChem, 2009, 10(11): 1757–1760. doi: 10.1002/cphc.v10:11 [48] CASIDA M E, GUTIERREZ F, GUAN J, et al. Charge-transfer correction for improved time-dependent local density approximation excited-state potential energy curves: analysis within the two-level model with illustration for H2 and LiH [J]. The Journal of Chemical Physics, 2000, 113(17): 7062–7071. doi: 10.1063/1.1313558 [49] HU C, SUGINO O, MIYAMOTO Y. Modified linear response for time-dependent density-functional theory: application to Rydberg and charge-transfer excitations [J]. Physical Review A, 2006, 74(3): 032508. doi: 10.1103/PhysRevA.74.032508 [50] HU C, SUGIN O. Average excitation energies from time-dependent density functional response theory [J]. Journal of Chemical Physics, 2007, 126: 074112. doi: 10.1063/1.2436887 [51] TAWADA Y, TSUNEDA T, YANAGISAWA S, et al. A long-range-corrected time-dependent density functional theory [J]. The Journal of Chemical Physics, 2004, 120(18): 8425–8433. doi: 10.1063/1.1688752 [52] TOKURA S, TSUNEDA T, HIRAO K. Long-range-corrected time-dependent density functional study on electronic spectra of five-membered ring compounds and free-base porphyrin [J]. Journal of Theoretical and Computational Chemistry, 2006, 5(4): 925–944. doi: 10.1142/S0219633606002684 [53] VYDROV O A, SCUSERIA G E. Assessment of a long-range corrected hybrid functional [J]. The Journal of Chemical Physics, 2006, 125(23): 234109. doi: 10.1063/1.2409292 [54] LIVSHITS E, BAER R. A well-tempered density functional theory of electrons in molecules [J]. Physical Chemistry Chemical Physics, 2007, 9(23): 2932–2941. doi: 10.1039/b617919c [55] PEACH M J G, TELLGREN E I, SAŁEK P, et al. Structural and electronic properties of polyacetylene and polyyne from hybrid and coulomb-attenuated density functionals [J]. The Journal of Physical Chemistry A, 2007, 111(46): 11930–11935. doi: 10.1021/jp0754839 [56] LIVSHITS E, BAER R. A density functional theory for symmetric radical cations from bonding to dissociation [J]. The Journal of Physical Chemistry A, 2008, 112(50): 12789–12791. doi: 10.1021/jp803606n [57] STEIN T, KRONIK L, BAER R. Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory [J]. Journal of the American Chemical Society, 2009, 131(8): 2818–2820. doi: 10.1021/ja8087482 [58] MAITRA N T, ZHANG F, CAVE R J, et al. Double excitations within time-dependent density functional theory linear response [J]. The Journal of Chemical Physics, 2004, 120(13): 5932–5937. doi: 10.1063/1.1651060 [59] GRITSENKO O V, BAERENDS E J. Double excitation effect in non-adiabatic time-dependent density functional theory with an analytic construction of the exchange–correlation kernel in the common energy denominator approximation [J]. Physical Chemistry Chemical Physics, 2009, 11(22): 4640–4646. doi: 10.1039/b903123e [60] ROMANIELLO P, SANGALLI D, BERGER J A, et al. Double excitations in finite systems [J]. The Journal of Chemical Physics, 2009, 130(4): 044108. doi: 10.1063/1.3065669 [61] SANGALLI D, ROMANIELLO P, ONIDA G, et al. Double excitations in correlated systems: a many–body approach [J]. The Journal of Chemical Physics, 2011, 134(3): 034115. doi: 10.1063/1.3518705 [62] SÄKKINEN N, MANNINEN M, VAN LEEUWEN R. The Kadanoff–Baym approach to double excitations in finite systems [J]. New Journal of Physics, 2012, 14(1): 013032. doi: 10.1088/1367-2630/14/1/013032 [63] HUIX-ROTLLANT M, IPATOV A, RUBIO A, et al. Assessment of dressed time-dependent density-functional theory for the low-lying valence states of 28 organic chromophores [J]. Chemical Physics, 2011, 391(1): 120–129. doi: 10.1016/j.chemphys.2011.03.019 [64] FURCHE F, AHLRICHS R. Absolute configuration of D2-symmetric fullerene C84 [J]. Journal of the American Chemical Society, 2002, 124(15): 3804–3805. doi: 10.1021/ja012207d [65] LIU J, LIANG W Z. Molecular-orbital-free algorithm for the excited-state force in time-dependent density functional theory [J]. The Journal of Chemical Physics, 2011, 134(4): 044114. doi: 10.1063/1.3548063 [66] LEVINE B G, KO C, QUENNEVILLE J, et al. Conical intersections and double excitations in time-dependent density functional theory [J]. Molecular Physics, 2006, 104(5/6/7): 1039–1051. [67] TAPAVICZA E, TAVERNELLI I, ROTHLISBERGER U, et al. Mixed time-dependent density-functional theory/classical trajectory surface hopping study of oxirane photochemistry [J]. The Journal of Chemical Physics, 2008, 129(12): 124108. doi: 10.1063/1.2978380 [68] KADUK B, VAN VOORHIS T. Communication: conical intersections using constrained density functional theory–configuration interaction [J]. Journal of Chemical Physics, 2010, 133: 061102. doi: 10.1063/1.3470106 [69] MAITRA N T. On correlated electron-nuclear dynamics using time-dependent density functional theory [J]. The Journal of Chemical Physics, 2006, 125(1): 014110. doi: 10.1063/1.2210471 [70] GONZE X, GHOSEZ P, GODBY R W. Density-polarization functional theory of the response of a periodic insulating solid to an electric field [J]. Physical Review Letters, 1995, 74(20): 4035. doi: 10.1103/PhysRevLett.74.4035 [71] GHOSEZ P, GONZE X, GODBY R W. Long-wavelength behavior of the exchange-correlation kernel in the Kohn-Sham theory of periodic systems [J]. Physical Review B, 1997, 56(20): 12811–12817. doi: 10.1103/PhysRevB.56.12811 [72] ONIDA G, REINING L, RUBIO A. Electronic excitations: density-functional versus many-body Green’s-function approaches [J]. Reviews of Modern Physics, 2002, 74(2): 601–659. doi: 10.1103/RevModPhys.74.601 [73] BOTTI S, SOTTILE F, VAST N, et al. Long-range contribution to the exchange-correlation kernel of time-dependent density functional theory [J]. Physical Review B, 2004, 69(15): 155112. doi: 10.1103/PhysRevB.69.155112 [74] SHARMA S, DEWHURST J K, SANNA A, et al. Bootstrap approximation for the exchange-correlation kernel of time-dependent density-functional theory [J]. Physical Review Letters, 2011, 107(18): 186401. doi: 10.1103/PhysRevLett.107.186401 [75] SHARMA S, DEWHURST J K, SANNA A, et al. Enhanced excitonic effects in the energy loss spectra of LiF and Ar at large momentum transfer [J]. New Journal of Physics, 2012, 14(5): 053052. doi: 10.1088/1367-2630/14/5/053052 [76] TANCOGNE-DEJEAN N, MÜCKE O D, KÄRTNER F X, et al. Impact of the electronic band structure in high-harmonic generation spectra of solids [J]. Physical Review Letters, 2017, 118(8): 087403. doi: 10.1103/PhysRevLett.118.087403 [77] BACZEWSKI A D, SHULENBURGER L, DESJARLAIS M P, et al. X-ray thomson scattering in warm dense matter without the chihara decomposition [J]. Physical Review Letters, 2016, 116(11): 115004. doi: 10.1103/PhysRevLett.116.115004 [78] MO C, FU Z, KANG W, et al. First-principles estimation of electronic temperature from X-ray thomson scattering spectrum of isochorically heated warm dense matter [J]. Physical Review Letters, 2018, 120(20): 205002. doi: 10.1103/PhysRevLett.120.205002 [79] SPERLING P, GAMBOA E J, LEE H J, et al. Free-electron X-ray laser measurements of collisional-damped plasmons in isochorically heated warm dense matter [J]. Physical Review Letters, 2015, 115(11): 115001. doi: 10.1103/PhysRevLett.115.115001 [80] RAMAKRISHNAN R, HARTMANN M, TAPAVICZA E, et al. Electronic spectra from TDDFT and machine learning in chemical space [J]. The Journal of Chemical Physics, 2015, 143(8): 084111. doi: 10.1063/1.4928757 [81] FUKS J I, NIELSEN S E B, RUGGENTHALER M, et al. Time-dependent density functional theory beyond Kohn–Sham Slater determinants [J]. Physical Chemistry Chemical Physics, 2016, 18(31): 20976–20985. doi: 10.1039/C6CP00722H [82] NIELSEN S E B, RUGGENTHALER M, VAN LEEUWEN R. Many-body quantum dynamics from the density [J]. EPL (Europhysics Letters), 2013, 101(3): 33001. doi: 10.1209/0295-5075/101/33001 [83] ELLIOTT P, MAITRA N T. Propagation of initially excited states in time-dependent density-functional theory [J]. Physical Review A, 2012, 85(5): 052510. doi: 10.1103/PhysRevA.85.052510 [84] CASTRO A, MARQUES M A L, ALONSO J A, et al. Excited states dynamics in time-dependent density functional theory [J]. The European Physical Journal D, 2004, 28(2): 211–218. doi: 10.1140/epjd/e2003-00306-3 [85] ALONSO J L, ANDRADE X, ECHENIQUE P, et al. Efficient formalism for large-scale ab initio molecular dynamics based on time-dependent density functional theory [J]. Physical Review Letters, 2008, 101(9): 096403. doi: 10.1103/PhysRevLett.101.096403 [86] MENG S, KAXIRAS E. Real-time, local basis-set implementation of time-dependent density functional theory for excited state dynamics simulations [J]. The Journal of Chemical Physics, 2008, 129(5): 054110. doi: 10.1063/1.2960628 [87] MENG S, KAXIRAS E. Mechanisms for ultrafast nonradiative relaxation in electronically excited eumelanin constituents [J]. Biophysical Journal, 2008, 95(9): 4396–4402. doi: 10.1529/biophysj.108.135756 [88] TULLY J C. Molecular dynamics with electronic transitions [J]. The Journal of Chemical Physics, 1990, 93(2): 1061–1071. doi: 10.1063/1.459170 [89] TAPAVICZA E, TAVERNELLI I, ROTHLISBERGER U. Trajectory surface hopping within linear response time-dependent density-functional theory [J]. Physical Review Letters, 2007, 98(2): 023001. doi: 10.1103/PhysRevLett.98.023001 [90] TAVERNELLI I, CURCHOD B F E, ROTHLISBERGER U. Mixed quantum-classical dynamics with time-dependent external fields: a time-dependent density-functional-theory approach [J]. Physical Review A, 2010, 81(5): 052508. doi: 10.1103/PhysRevA.81.052508 [91] KRISHTAL A, CERESOLI D, PAVANELLO M. Subsystem real-time time dependent density functional theory [J]. The Journal of Chemical Physics, 2015, 142(15): 154116. doi: 10.1063/1.4918276 [92] PAVANELLO M. On the subsystem formulation of linear-response time-dependent DFT [J]. The Journal of Chemical Physics, 2013, 138(20): 204118. doi: 10.1063/1.4807059 [93] NEUGEBAUER J. Couplings between electronic transitions in a subsystem formulation of time-dependent density functional theory [J]. The Journal of Chemical Physics, 2007, 126(13): 134116. doi: 10.1063/1.2713754 -
图( 7) 表( 1)
计量
- 文章访问数: 13766
- HTML全文浏览数: 13766
- PDF下载数: 499
- 施引文献: 0