
 首页
首页 登录
登录 注册
注册




 下载:
下载:


-
橄榄石在上地幔和俯冲带中的质量分数超过50%,是俯冲带和上地幔中最主要的组分[1-2]。研究橄榄石相变过程不仅可以了解地球内部特征,还有助于揭示地震发生机制。板块俯冲是两个构造板块汇聚时在其边界上发生的、伴有板块消亡的地质过程,是板块构造理论的重要内容之一。每年有80~120 km2的岩石圈层俯冲到350 km以下的界面,并在俯冲过程中被逐渐加压加热[3]。由于下降岩石板块既厚又是热的不良导体,故能在俯冲过程中保持较冷的状态。在正常地幔条件下,α相橄榄石在大约410 km 深度的温压条件下变得不稳定,开始转变为β相,标志着地幔转换带的开始[4];在约520 km 深度的温压条件下,β相橄榄石转变为γ相橄榄石[5];在约 660 km 深度处,γ相橄榄石分解成钙钛矿和镁方铁矿,标志着下地幔的开始[4]。但是在岩石圈俯冲板块内,由于俯冲岩石层内部温度低,在约 400 km 的深度α相橄榄石不发生相变,低压相的橄榄石以亚稳态的形式进入更深的深度[3]。Sung 等[6]用金刚石压腔研究了天然组分橄榄石粉末样品,认为在低于973 K的较冷俯冲板块内部,橄榄石处于亚稳态。韩鹏等[7]研究发现,俯冲带的板块边界内板块运动与地震各向异性及应力场等存在相关性。刘雷等[3]提出,在地球深部岩石脆性行为失效的情况下,橄榄石等矿物的亚稳态相变可能是诱发深源地震的因素之一,在亚稳态条件下橄榄石等矿物发生相变会包含一个小的体积变化,形成一个狭长弱剪切带,同时快速释放大量剪切能,即相变破裂诱发深源地震的机制。
对橄榄石高温高压结构及物理性质的实验研究主要采用静高压方法[5, 8-11]。1962年,Bragg和Brown测定了橄榄石的晶体结构,空间群为Pbnm(α相),斜方晶系,化学通式为(M(1)M(2))2SiO4,对于天然橄榄石,M代表Mg2+、Fe2+且完全类质同相[12]。β相和γ相是橄榄石的高温高压相,空间群分别为Imma和
$Fd\bar 3m$ [13-14]。关于亚稳态的橄榄石,马艳梅等[12, 15]利用同步辐射X射线衍射和拉曼光谱研究发现,在常温高压(低于40 GPa)和常压高温(377~1 113 K)两种条件下,α相橄榄石结构均稳定,未发生由压力或温度导致的相变。在实际的俯冲板块和上地幔地质环境中,矿物脱水反应和板块俯冲带入的水是大量存在的[16-18],一般认为水对橄榄石相变过程产生很大的影响[19-23]。Hosoya等[24]和Kubo等[25]研究了水对亚稳态橄榄石存在范围的影响,结果表明,Mg2SiO4橄榄石α-β相变速率与OH含量成正比,并且水含量的增加会大大降低亚稳态橄榄石的存在范围。本研究结合拉曼光谱和同步辐射X射线衍射(X-ray diffraction, XRD)技术,对河北阳原捕虏体中的橄榄石进行较低温度下的快速压缩实验,探索快速压缩是否促进橄榄石结构相变和脱水等反应,为研究地震发生瞬间橄榄石的结构变化提供参考。
全文HTML
-
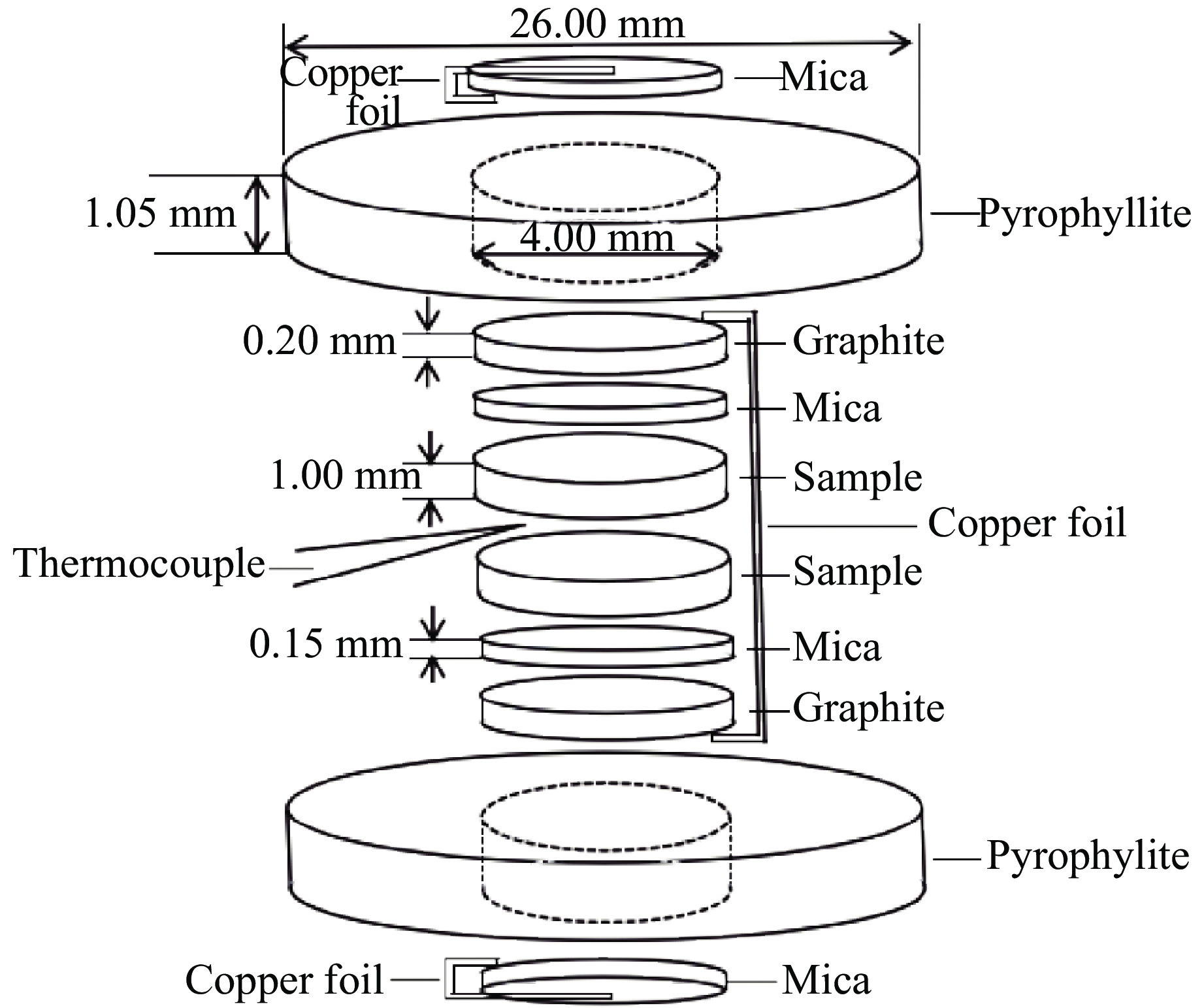
采用河北阳原玄武岩包裹体中的橄榄岩,利用高倍显微镜挑选出透明无杂质的新鲜橄榄石。高压实验在快速增压装置[26]上进行,高压模具为硬质合金平面对顶压砧,砧面直径为26 mm,样品组装如图1所示。快速加压过程中样品压力上升时间约为120 ms,样品压力由铋相变标定的压机油压与样品腔实际压力的关系推算得到[27]。受快速增压实验装置(含油压系统、压力和温度传感器等)自身精度的限制,压力测量结果存在误差。在相同设置下多次重复实验数据表明,快速压缩引起的压力增幅的相对误差约为4.4%[27]。本实验主要分两部分。第一部分是将研磨后的粉末初始样品放入高压模具中,将5个样品分别加热到373、473、573、673、773 K时,在约120 ms的时间内快速加压至3 GPa,加压完成后随即切断加热电路降温,由于压砧等的导热性良好,样品温度瞬间下降至室温,随后缓慢卸压并回收样品。样品温度通过夹在两片样品中间的K型热电偶及温度记录仪(Graphtec GL900)测量,误差约为0.1 K。第二部分是将研磨后的粉末初始样品放入马弗炉中,分别在873、973、1073、1 173 K下焙烧2 h,将焙烧后的橄榄石样品放入高压模具中,在室温下快速加压至3 GPa,卸压并回收样品。
对回收样品进行同步辐射XRD、拉曼光谱和红外吸收光谱测试,并通过扫描电镜(Scanning electron microscope, SEM)观察微观形貌,分析快速加压过程对橄榄石晶体结构及分子振动模式的影响。研磨后的初始样品和回收样品的拉曼光谱测量采用光谱分辨率为1.0 cm–1的背散射拉曼光谱仪(in-Via, Renishaw),激发光源采用波长为532 nm的单色光,拉曼光谱测量范围为0~1 000 cm–1。研磨后的橄榄石初始样品的常规XRD分析(X’Pert.PRO.MPD.Philips)采用Cu靶Kα激发线,波长1.54 Å。研磨后的初始样品和回收样品的同步辐射XRD实验在上海同步辐射光源(Shanghai Synchrotron Radiation Facility, SSRF)的高压衍射线站15 U1完成。该线站所使用的能量为20 keV,X射线衍射波长为0.619 9 Å。实验过程中光斑尺寸(半高宽)为2 μm × 3 μm。采用面探测器收集衍射信息,然后使用FIT2D软件积分得到衍射曲线[28]。采用扫描电镜(FEI Inspect F50)观察研磨后的初始样品和回收样品微观形貌。采用傅里叶变换红外光谱(Nicolet iS10)对室温快压、1 073 K焙烧后快压至3 GPa、升温773 K后快压至3 GPa的橄榄石回收样品进行红外光谱分析。
-
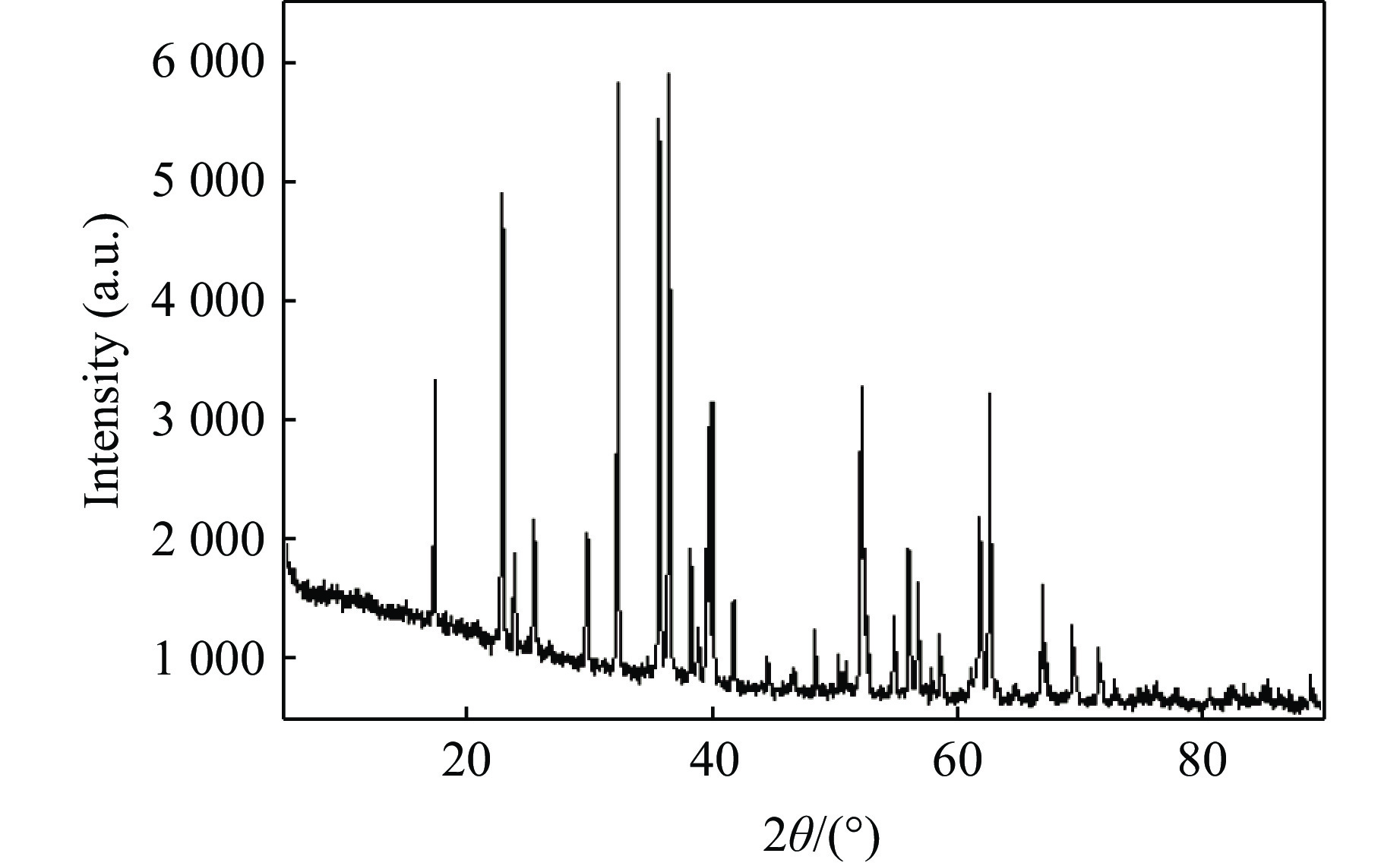
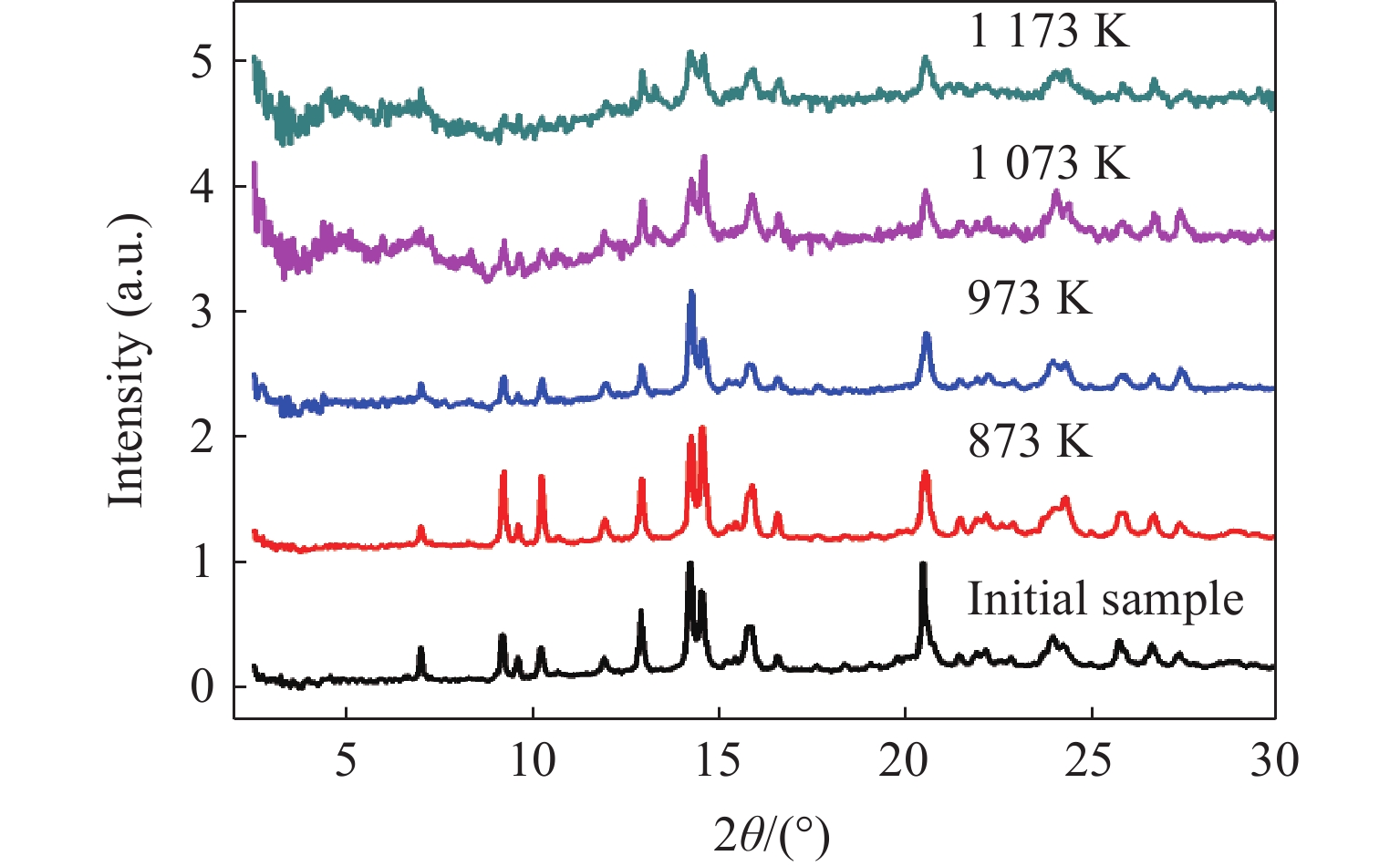
图2是橄榄石初始样品的常温常压XRD谱,与Mg2SiO4标准谱比较,确定橄榄石样品的晶体结构为α相,即空间群为Pbnm。橄榄石初始样品的晶胞参数:a = 5.982 Å,b = 10.198 Å,c = 4.755 Å。通过SEM能谱分析,测得初始样品中Mg和Fe的质量分数分别约为32.0%和6.5%,化学式为(Mg0.91Fe0.09)2SiO4。快速压缩实验回收的橄榄石样品量少,结晶性变差,衍射信号弱且有展宽,细聚焦同步辐射X射线的能量高、波长短、分辨率高,有助于分析橄榄石回收样品的微观结构。图3和图4分别为升温快压和焙烧快压后回收的橄榄石同步辐射XRD谱。与初始样品的XRD谱对比可得,在升温快压(上升温度373~773 K,快压至3 GPa)和焙烧后室温快压(焙烧温度873~1 173 K,快压至3 GPa)两种实验条件下,橄榄石衍射峰的位置并未发生改变,也没有出现新的衍射峰,说明在上述过程中α相橄榄石晶体的结构稳定,未发生因快速压缩、升温或焙烧共同作用而导致的α-β相变。
图5显示了由SEM观察到的不同条件下回收橄榄石样品的微观形貌。图5(a)和图5(b)中,初始样品和室温(293 K)下快压后的橄榄石样品中都包含较大的块状结构,且表面较光滑。图5(c)~图5(f)中,在升温快压和焙烧后快压两种条件下,橄榄石表面更加粗糙,附着较小的颗粒,并且颗粒间距变小,推测是由温度和压力共同作用而导致的,在较高温度下或者较高温度焙烧后橄榄石的硬度降低,可碎性提高,快速压缩过程使橄榄石颗粒细化。
-
橄榄石是岛状的硅酸盐矿物,其晶体结构是不共用氧原子的,晶体结构中孤立的SiO4四面体彼此不直接连接,由位于八面体空隙中的阳离子(Mg2+、Fe2+)连接起来,属于Td对称[29]。然而由于在阳离子晶体场的作用下,硅氧四面体发生很大的畸变,所以其对称性降低为Cs对称。橄榄石的拉曼特征峰是由于Si—O键的振动造成的,拉曼特征峰的变化反映了橄榄石硅氧四面体中Si—O键的振动变化。基于简并的原因,硅氧四面体的振动模式在0~1 000 cm–1波数范围内被分为:高于800 cm–1 的Si—O 伸缩振动模(对称伸缩振动模ν1和反对称伸缩振动模ν3)、400~700 cm–1的Si—O 弯曲振动模(对称弯曲振动模ν2和反对称弯曲振动模ν4)、低于400 cm–1的旋转振动模(R)和金属-氧的平移振动模(T)。另外,橄榄石晶格振动光谱理论研究表明,硅氧四面体的振动模式共有84个,对称性分类为
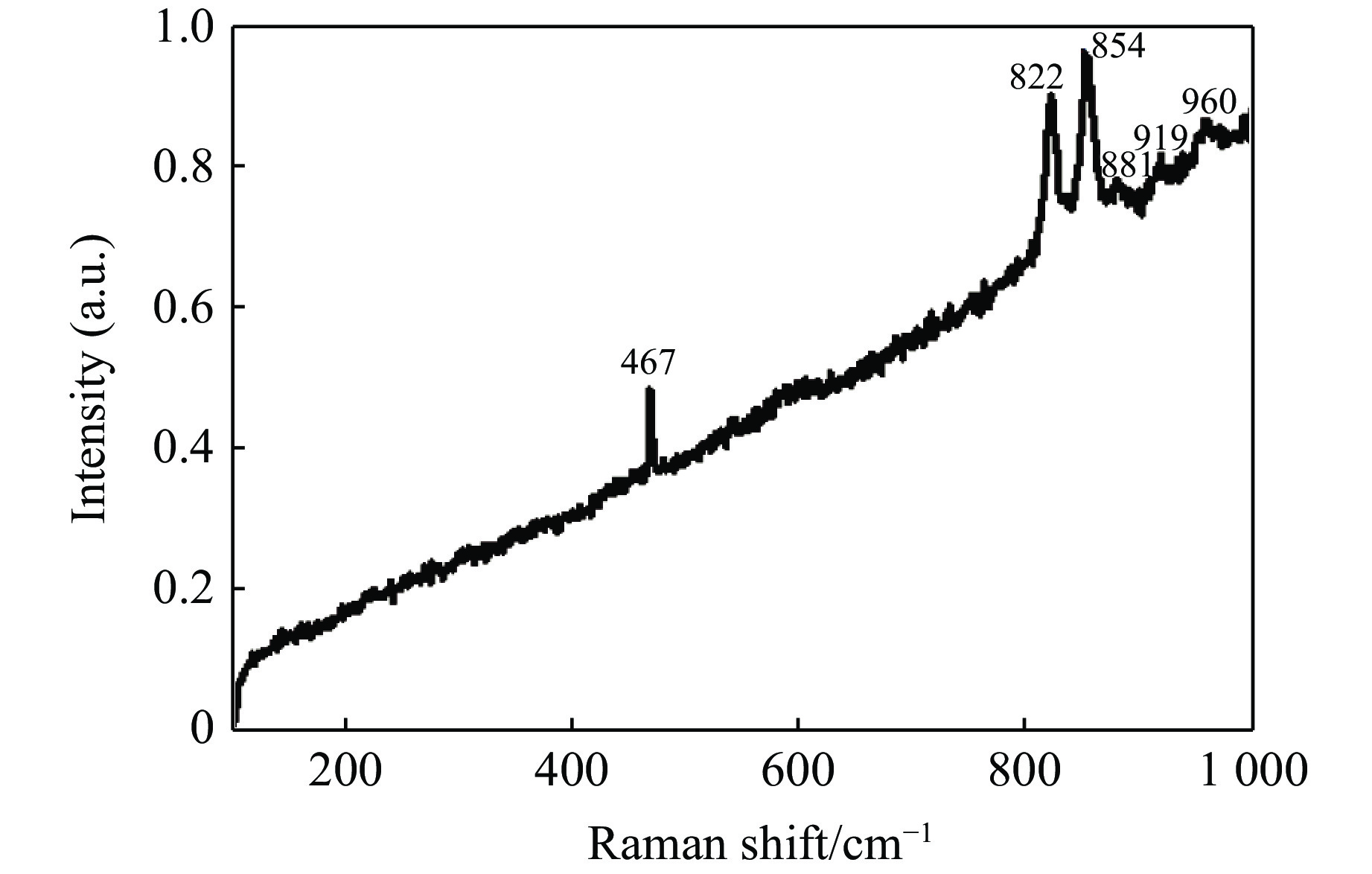
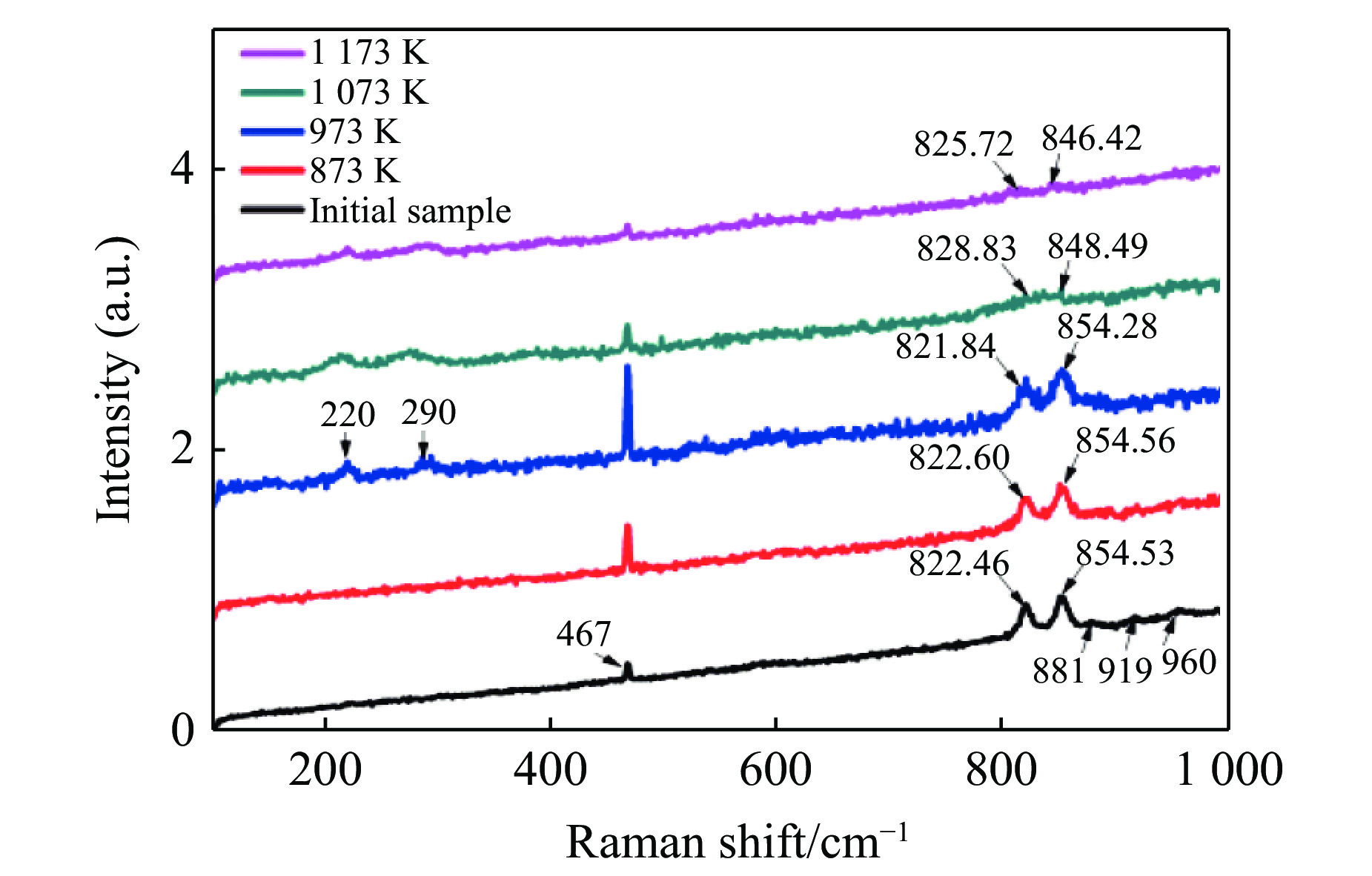
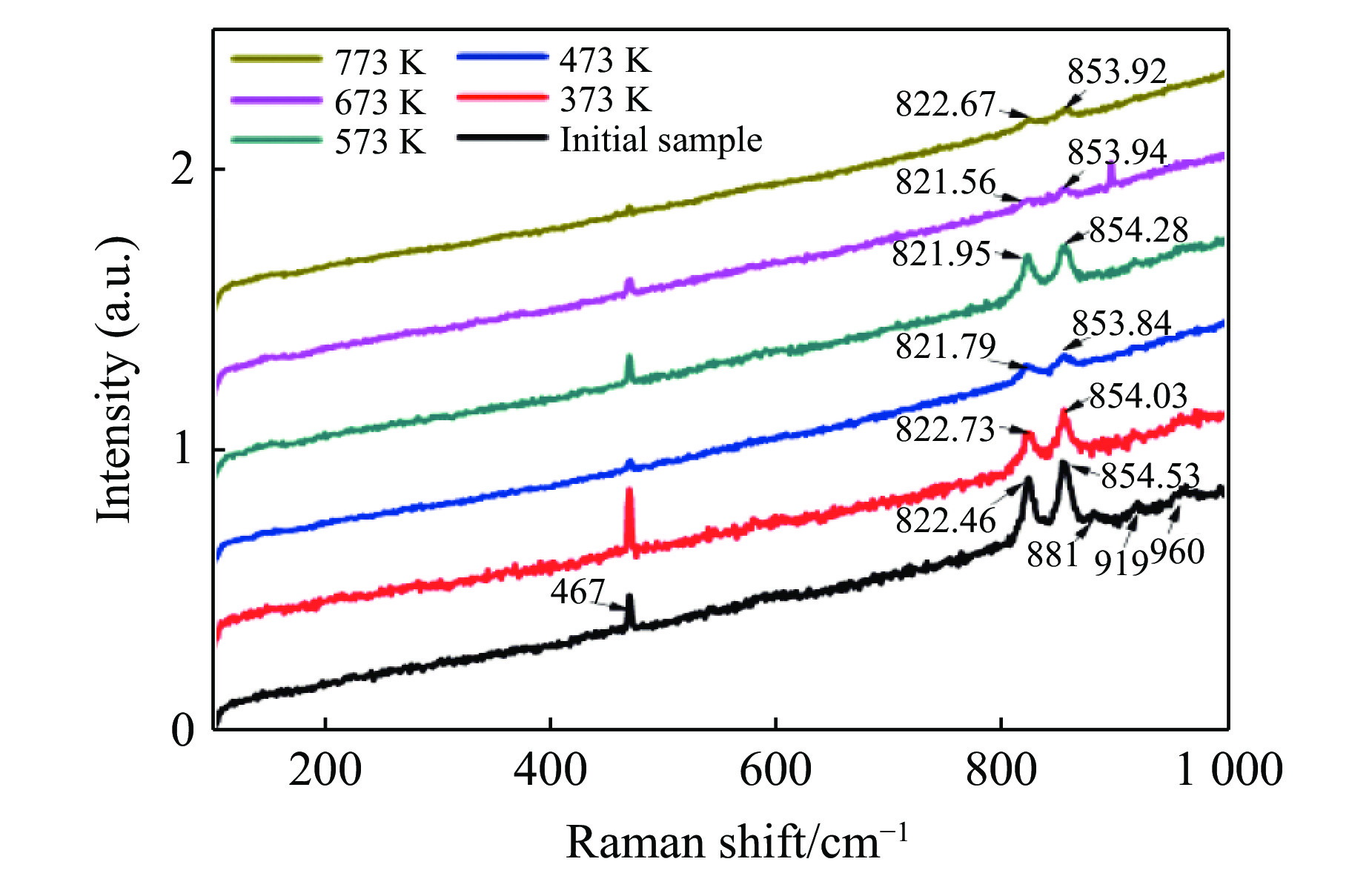
$11{A_{\rm g}} + 7{B_{1{\rm g}}} + 11{B_{2{\rm g}}} +$ $ 7{B_{3{\rm g}}} + 10{A_{\rm u}} + 14{B_{1{\rm u}}} + 10{B_{2{\rm u}}} + 14{B_{3{\rm u}}}$ ,其中具有拉曼振动活性的振动模式为${A_{\rm g}} + {B_{1{\rm g}}} + {B_{2{\rm g}}} + {B_{3{\rm g}}}$ ,共36个[9, 30]。橄榄石初始样品的常温常压拉曼光谱如图6所示,主要观测到467、822、854、881、919、960 cm–1 6个拉曼峰。在橄榄石的晶体结构中,硅氧四面体内部原子之间的键合力远远大于基团之间的结合力,而拉曼光谱中最强的两个拉曼特征峰822 cm–1和854 cm–1就来源于硅氧四面体的对称伸缩振动和反对称伸缩振动。将本实验中观测到的橄榄石拉曼光谱与文献中镁橄榄石的拉曼光谱进行了对比,如表1[30-31]所示。
通过分析橄榄石拉曼特征峰的变化可以获得Si—O键的振动模式变化。拉曼位移可以表示为
式中:c为光速,μ为折合质量,K为振动键的力常数。在共价键分子中
式中:N为阿伏伽德罗常数,r为A、B原子间的距离,a、b为常数,χA、χB为A、B原子的参数。对于硅氧四面体而言,c、μ为常数,由式(1)可以得到影响硅氧四面体拉曼位移的因素只有K值的大小。除此之外,原子A和B是确定的,通过式(2)分析可得,K值的大小只取决于A、B两原子间的距离r。因此,通过式(1)和式(2)可以分析得到橄榄石硅氧四面体中硅氧原子间距离的变化趋势。
图7为不同温度焙烧并在室温下快压后回收橄榄石的拉曼光谱。与橄榄石初始样品的拉曼光谱相比,873 K和973 K焙烧后快压回收样品的822 cm–1和854 cm–1振动峰的位置基本不变,1 073 K和1 173 K焙烧后快压回收样品的822 cm–1和854 cm–1振动峰强度明显变弱,并且822 cm–1振动峰向高波数移动,854 cm–1振动峰向低波数移动。李月等[32]的实验结果显示,α相橄榄石的822 cm–1和854 cm–1拉曼特征峰在室温下随压力的升高向高波数方向偏移,两个拉曼峰之间的波数差减小,半峰宽增大。α相橄榄石原位高温拉曼光谱研究发现,常压下822 cm–1和854 cm–1拉曼特征峰随着温度的增加向低波数方向移动。压力和温度单一变量作用主要改变Si—O键的键长。压力作用下,键长变短,力常数增加,拉曼振动峰移向高波数。温度作用下,晶体膨胀,键长变长,力常数减小,振动峰移向低波数[15]。上述变化在卸压或降温后通常都是可逆的[15,32]。本工作中,1 073 K以上温度焙烧并快压后振动峰位发生不可逆变化,可能与残余应力作用下硅氧四面体畸变有关。
图8为不同温度下快压后回收橄榄石的拉曼光谱。和高温焙烧后快压样品的拉曼光谱(见图7)相比,图8所示拉曼特征峰峰位未发生明显移动,间接说明1 073 K温度以上的高温经历对橄榄石的结构稳定性产生重要影响。值得注意的是,回收样品的467、881、919、960 cm–1拉曼峰强度随快压时样品温度上升而逐渐减小以致消失。同时,Si—O对称伸缩和反对称伸缩的混合振动模对应的拉曼峰(常温下位于822 cm–1和854 cm–1)展宽。推测这些变化与升温快压后样品晶格破坏有关,结合图5中的微观形貌发现,快压后样品颗粒变小、晶粒细化是拉曼峰展宽的主要原因。
-
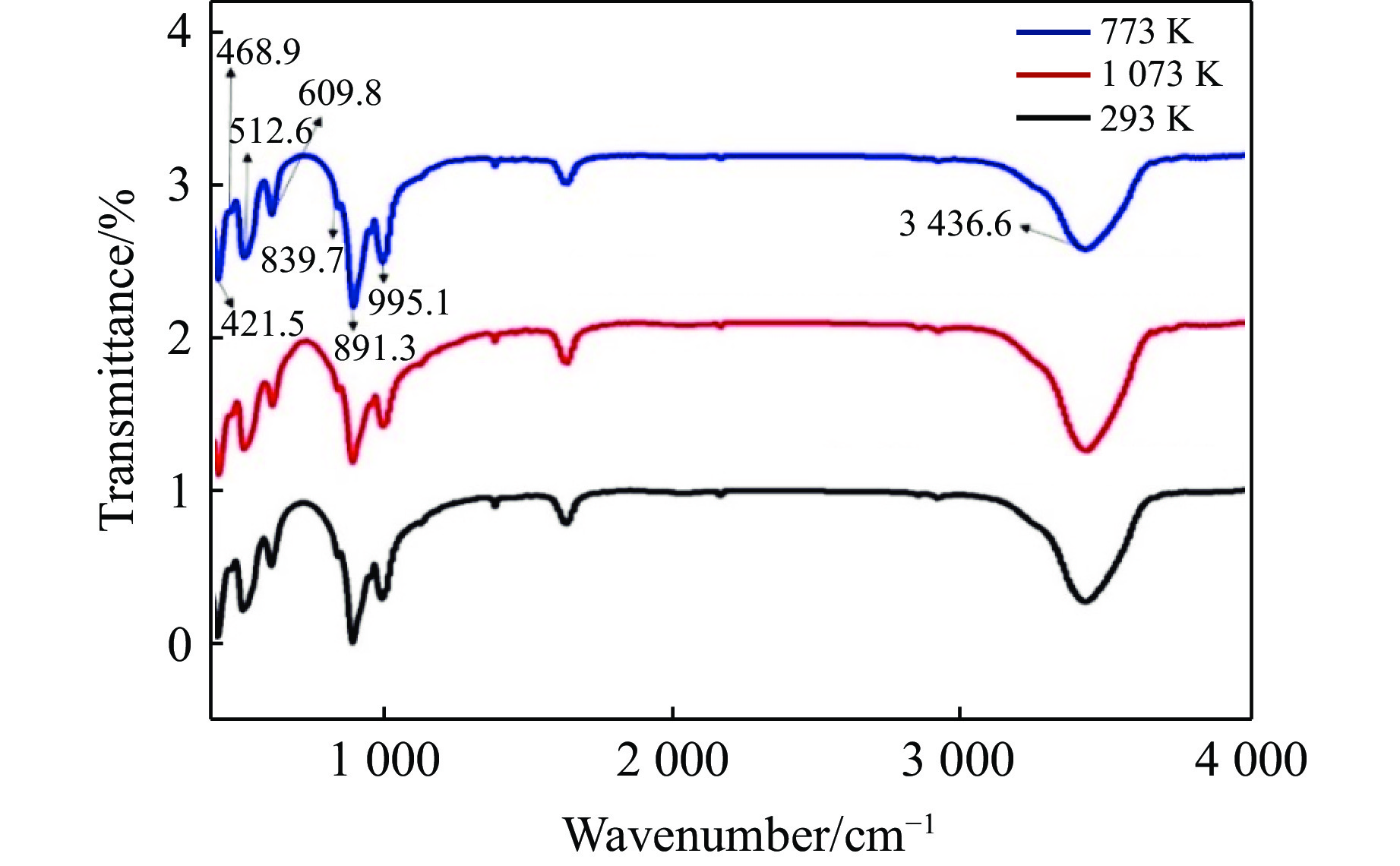
朱蓓蓓等[33]研究发现,阳原捕虏体中橄榄石结构水含量明显较高,可能与阳原橄榄岩经历的上地幔交代作用有关。扩散进入橄榄石中的水以羟基(质子H与硅酸盐骨干中的O原子结合)为主要存在形式,羟基的红外吸收峰主要位于3 200~3 620 cm–1波数[23]。
为了了解温度和快压共同作用对橄榄石样品中水含量的影响,对室温快压、焙烧1 073 K后室温快压、升温到773 K快压后回收的3个橄榄石样品研磨后进行红外光谱分析,光谱采集的波数范围为400~4 000 cm–1,光谱归一化后如图9所示。从图9中可以看到:3种实验条件下回收的橄榄石红外光谱基本一致。400~1 000 cm–1波数范围内的吸收峰主要是阳离子晶体场作用下的硅氧四面体振动吸收峰[9]。3 200~3 620 cm–1波数范围内羟基吸收峰的位置和相对强度均未发生明显的改变,推测较低压力和温度下的快速增压过程对橄榄石中结构水含量未产生明显影响。
2.1. 橄榄石晶体结构及微观形貌
2.2. 橄榄石分子振动模式
2.3. 橄榄石晶体的红外光谱
-
传统静高压实验的压力加载时间长,无法研究地震瞬间俯冲带内亚稳橄榄石的结构演变;冲击压缩实验的压力加载时间短,但常常伴随着温度的大幅升高,无法满足俯冲带较冷环境下亚稳橄榄石的快速压缩实验研究。介于静高压和动高压之间、加压时间为毫秒量级的快速压缩实验有利于研究地震发生瞬间亚稳态橄榄石的结构变化。对河北阳原捕虏体中的新鲜橄榄石在升温快压和焙烧后室温快压两种条件下的结构稳定性进行了研究,发现在873~1 173 K焙烧2 h后室温快压至3 GPa和在373~773 K升温快压至3 GPa的条件下,橄榄石均未发生由温度和快压共同作用所导致的相变,晶体结构保持稳定。尽管相比于俯冲带的温压条件,本实验的压力条件较低,但是通过实验仍然观察到了快压和温度共同作用下橄榄石可碎性提高、晶粒细化以及结构水稳定等现象,对于研究地震发生瞬间橄榄石的结构变化具有一定的参考价值。