
 首页
首页 登录
登录 注册
注册




 下载:
下载:

-
随着材料科学研究的深入,研究者们对材料的研究不再局限于常压范围,开始更多地关注高压下材料的结构和物理性质。在高压作用下,晶体的体积被压缩,原子间的间距变小,电子轨道发生变化,甚至原子的周期性排列也会改变,使晶体结构发生相变[1-3]。晶体结构在高压环境下具有较为丰富的相变序列,许多常压下无法合成的材料可在高压环境下合成,因此利用高压技术,可以丰富人们对物质世界的认识[4-5]。
高压下聚合氮以及氮化物的物理、化学性质受到较多关注。将氮作为媒介来合成不同结构的含氮材料是研究聚合氮及氮化物的基础[6-8],如利用氮单质在高温高压环境下合成立方氮[9],利用氮元素和其他元素合成具有超硬性质的化合物等[10]。在氮的化合物中,金属氮化物的潜在应用价值巨大,如超导材料[11]、特殊化学制品[12]、热稳材料、高能量密度材料和超硬材料等[13-14]。针对金属氮化物的研究也愈发广泛。
对于碱土金属氮化物的理论研究较多。Be3N2的晶体结构研究结果表明,R3m和
$P\bar 3m1$ 两个晶体结构的维氏硬度分别为51 GPa和54 GPa,说明两种结构的Be3N2是潜在的超硬材料[15]。在高压下发现Ca与N的多个配比结构,得出CaN3的C2/c结构中N原子形成孤立的六环;进一步对键长和键级进行计算,发现该结构的CaN3是潜在的高能量密度材料[16]。Mg3N2在高压下具有丰富的物理性质。研究发现,H2分子很容易在τ-Mg3N2、α-Mg3N2和α′-Mg3N2相的表面分解为H原子并进入材料内部,说明Mg3N2是潜在的储氢材料[17]。由上述研究可知,碱土金属氮化物的物理性质丰富,潜在应用价值巨大。为此,对高压下Mg的多氮化合物的晶体结构和物理性质进行研究具有十分重要的意义。本研究结合晶体结构预测技术和基于密度泛函理论的第一性原理方法,在0~100 GPa的压强范围内,对MgN8的晶体结构进行预测,并系统分析预测得到的α-MgN8、β-MgN8和γ-MgN8相,所得结论对进一步研究碱土金属氮化物具有一定的参考价值。
全文HTML
-
采用基于粒子群优化算法的CALYPSO软件,对MgN8进行结构搜索[18],压强范围为0~100 GPa,模拟晶胞采用2倍胞和4倍胞。对于搜索出的结构,使用VASP软件包进行结构优化[19],其中描述电子间的交换关联势能采用广义梯度近似(GGA)下的Perdew-Burke-Ernzerh(PBE)交换关联泛函[19],赝势采用全电子投影缀加平面波方法[20],Mg原子的价电子为2p63s2,N原子的价电子为2s22p3。为了保证计算精确,使能量收敛精度小于1 meV/atom,经过能量收敛测试,得到平面波的截断能为600 eV;第一布里渊区积分采用Monkhorst-Pack网格方法,网格的间距为0.2 nm–1;优化过程中,在确保空间群不改变的情况下,选用10–5 eV为自洽能量收敛的最小值,优化应力收敛设置为0.001 eV/Å。对于优化后的结构,使用超晶胞的方法,利用PHONOPY软件计算声子色散关系和声子态密度,选取不同的扩胞数和k点网格:α-MgN8相为1 × 1 × 1和7 × 7 × 6;β-MgN8相为2 × 2 × 4和6 × 6 × 9;γ-MgN8相为3 × 1 × 1和9 × 5 × 4。使用VASP软件包计算电子局域函数。
-
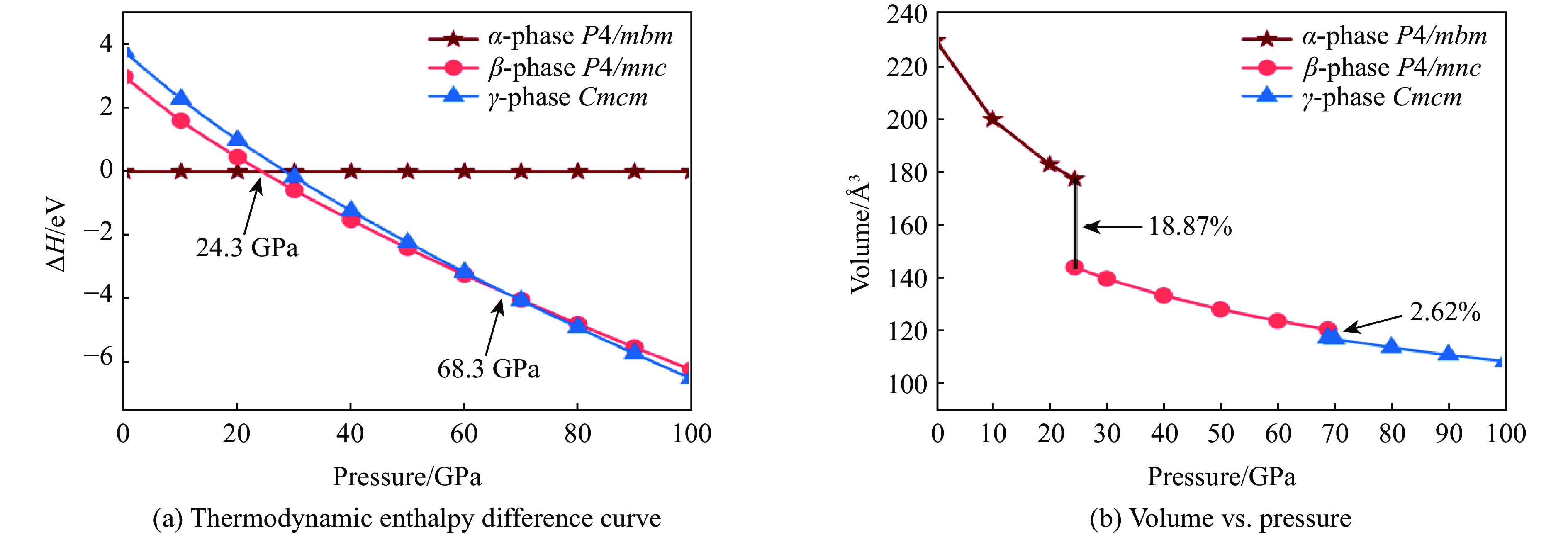
运用CALYPSO软件搜索,在0~100 GPa压强范围内,对预测出的MgN8晶体结构进行晶格常数和原子位置的优化,计算时设定的温度为0 K,根据G = H – TS(G为吉布斯自由能,H为焓,T为温度,S为熵),可以用焓代替系统的自由能。通过计算优化后结构的焓值随压强的变化关系,可以得到晶体结构的热力学稳定区间。绘制出的焓差随压强变化的曲线如图1所示。由图1(a)可知:在常压下,空间群为P4/mbm的MgN8晶体结构的焓值最低,记为α-MgN8相;当压强达到24.3 GPa时,空间群为P4/mnc的晶体结构具有更低的焓值,记为β-MgN8相;当压强达到68.3 GPa时,空间群为Cmcm的晶体结构能量更低,记为γ-MgN8相。图1(b)给出了α-MgN8、β-MgN8和γ-MgN8相的晶胞体积随压强的变化。分析可知:α-MgN8相的晶胞体积随着压强的增大而减小,晶胞体积随压强连续变化;当压强达到24.3 GPa时,MgN8发生体积坍缩,坍缩率为18.87%;在68.3 GPa时,发生结构相变,体积塌缩率为2.62%;两次相变均属于一级相变。
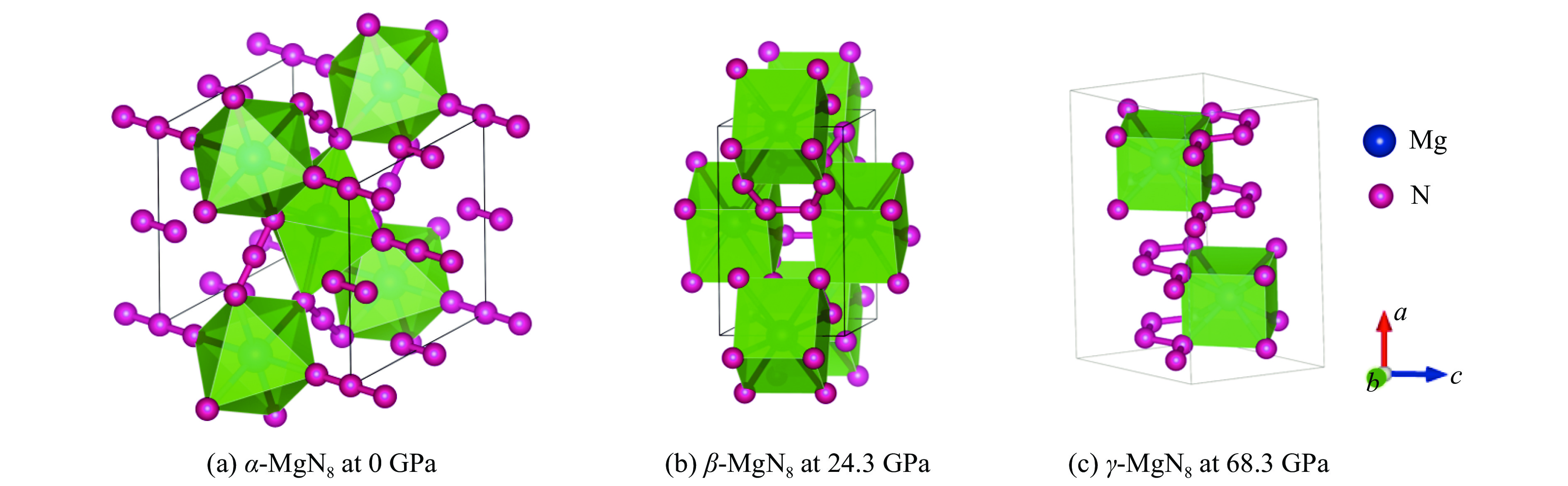
α-MgN8、β-MgN8和γ-MgN8相的晶体结构见图2,其平衡态晶格常数以及原子位置信息列于表1和表2。
图2(a)所示的结构为0 GPa下优化后的α-MgN8相结构。在α-MgN8相结构中,每个Mg原子被6个N原子包围,构成八面体结构。其中,Mg-N的键长有2类:一类是中心Mg原子与八面体上下顶点的两个N相连,键长为2.280 Å;另一类为中心的Mg原子与八面体中其余4个N相连,键长为2.109 Å。α-MgN8相的晶格结构常数:a = b = 5.913 Å,c = 6.572 Å,α = β = γ = 90.0°。其中,Mg原子的Wyckoff占位为2b (0.000, 0.000, 0.500),N原子的Wyckoff占位分别为2a (0.000, 0.000, 0.000)、2c (0.000, 0.500, 0.500)、4e (0.000, 0.000, 0.821)、4f (0.000, 0.500, 0.915)和4h (0.859, 0.359, 0.500)。图2(b)所示的结构为24.3 GPa下优化后的β-MgN8相结构。在该结构中,每个Mg原子被8个N原子包围,8支Mg—N键的键长相同,为2.184 Å,构成正六面体。该结构的晶体结构常数:a = b = 6.219 Å,c = 3.724 Å,α = β = γ = 90.0°。其中,Mg原子的Wyckoff占位为2a (0.000, 0.000, 1.000),N原子Wyckoff占位为16i (0.416, 0.219, 0.823)。图2(c)所示的结构为68.3 GPa下优化后的γ-MgN8相结构。在该结构中,每个Mg原子与8个N原子相连,构成六面体结构。六面体中的8支Mg-N键有2种键长:4支2.081 Å和4支2.069 Å。该结构的晶体结构常数:a = 4.167 Å,b = 4.167 Å,c = 8.680 Å,α = β = 90.0°,γ = 51.1°。其中,Mg原子的Wyckoff占位为8g (0.624, –0.376, 1.250),N原子Wyckoff占位分别为16h (0.226, –0.135, 0.617)和16h (1.115, 0.471, 1.386)。
-
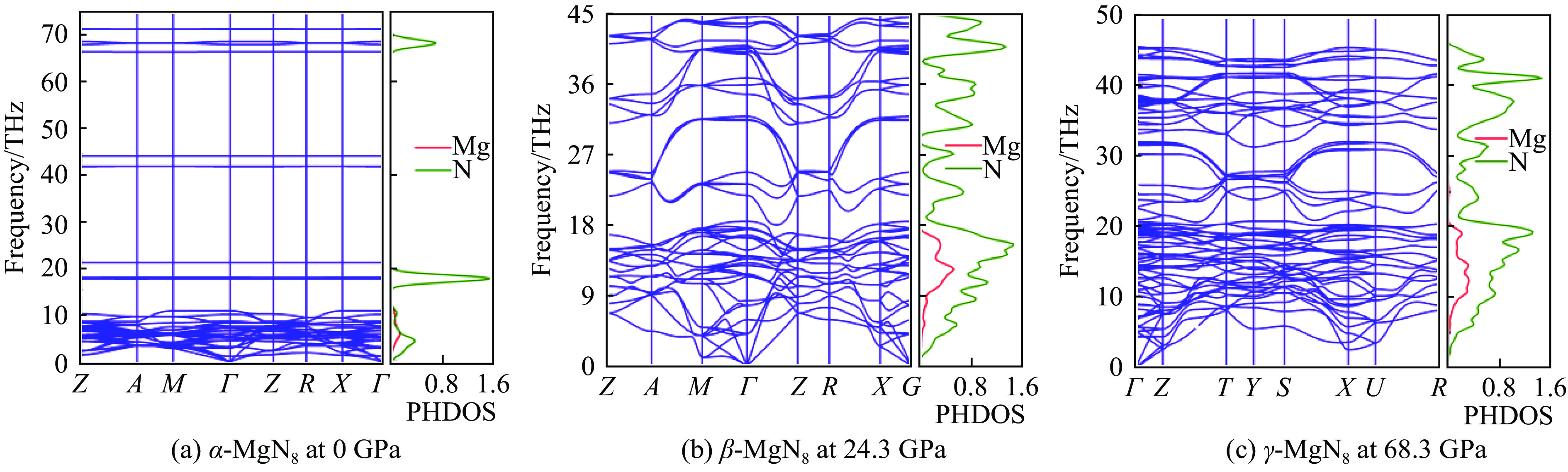
通过计算声子色散关系,可以判断晶格结构是否具有动力学稳定性。晶格结构具有动力学稳定性的判断条件是,所有简正声子频率都为有限的实值[21],如果是虚值,就可以判断此结构出现了声子软化现象,晶格结构不稳定。计算了预测结构的声子谱和声子态密度,如图3所示。
由图3(a)可知,0 GPa时α-MgN8相在整个布里渊区未出现声子软化现象,说明该相具有动力学稳定性,最大光学支的频率为71.35 THz。由图3(b)和图3(c)可知,24.3 GPa和68.3 GPa时,β-MgN8和γ-MgN8相具有动力学稳定性,最大光学支频率分别为44.64 THz和45.63 THz。
-
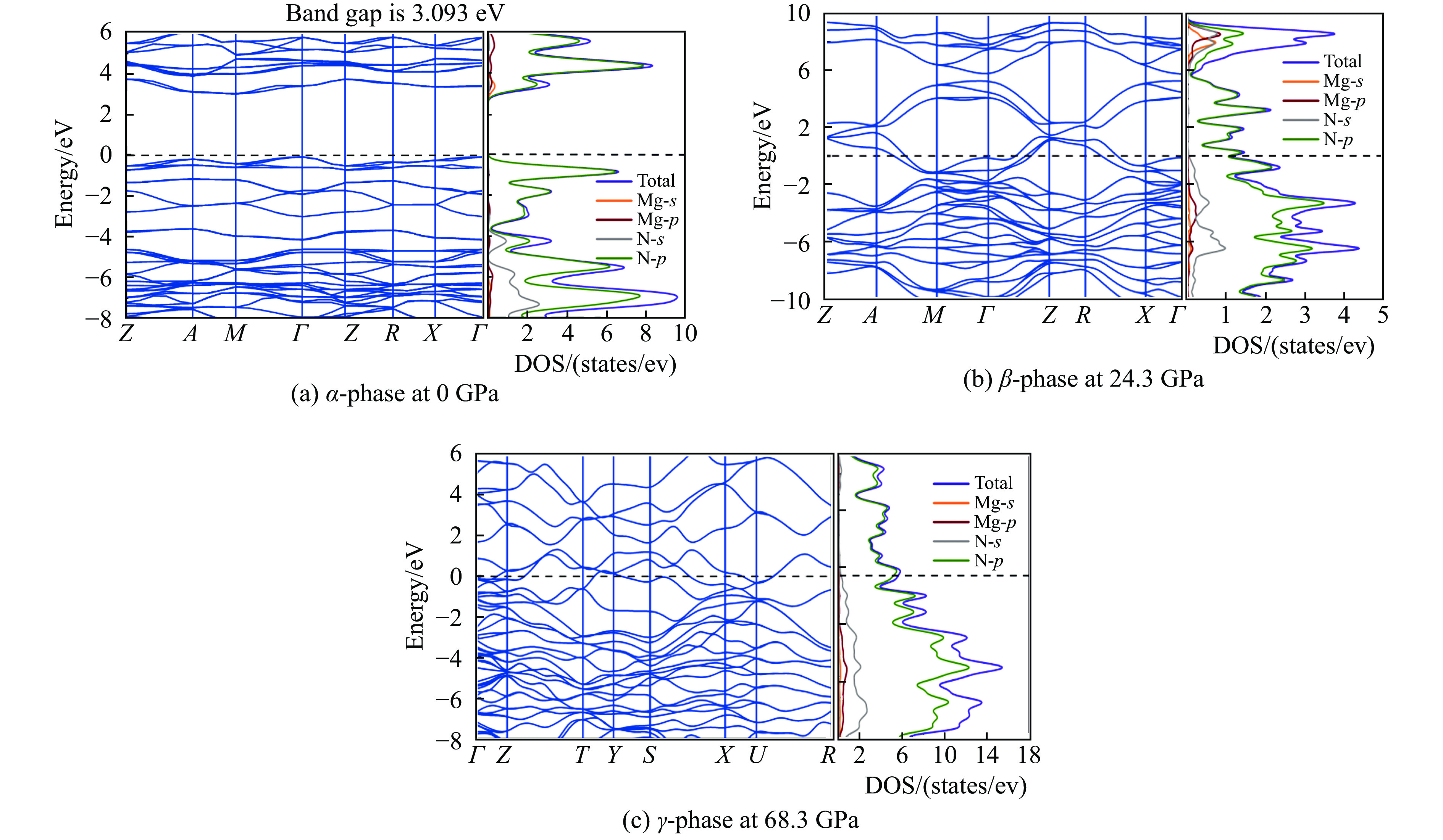
为了探究预测得到的MgN8晶体结构的电子性质,分别研究了α-MgN8相在0 GPa、β-MgN8相在24.3 GPa和γ-MgN8相在68.3 GPa时的能带结构和电子态密度,如图4所示。
由能带图可知,压强为0 GPa时,α-MgN8相的导带与价带之间有3.093 eV的带隙,表明该相结构具有非金属性;当压强为24.3 GPa和68.3 GPa时,β-MgN8和γ-MgN8相的导带与价带跨越费米面发生交叠,表明这两相结构具有金属性。
由电子态密度的计算结果可知,α-MgN8、β-MgN8和γ-MgN8相费米能级处的电子态密度均主要由N原子的p轨道贡献,而N原子的s轨道和Mg原子的s、p轨道对电子态密度的贡献相对较少。N原子的p轨道与s轨道之间存在杂化,形成了N—N共价键。
为了研究α-MgN8、β-MgN8和γ-MgN8相晶体结构的化学键,计算了这3个相的电子局域函数(Electron localization function, ELF)[22],如图5所示。
图5(a)显示了α-MgN8结构的三维电子局域函数。α-MgN8相的N原子之间形成N2和N3,其中:N2的两个N原子之间存在电子局域,形成了共价键,键长为1.116 Å,两个N原子外围存在孤对电子;N3中间的N原子与相邻的两个N原子之间存在电子局域,形成两种N3共价键,其键长分别为1.178 Å和1.182 Å,N3两侧的N原子周围也存在孤对电子。这3种N-N键的键长介于N≡N三键(1.10 Å)和N=N双键(1.25 Å)之间。图5(b)显示了压强为24.3 GPa时β-MgN8结构的三维电子局域函数。该相N原子之间形成链状结构,每个N原子与相邻的两个N原子之间均有电子局域,形成共价键,键长为1.308 Å和1.316 Å,这两种N-N键的键长介于N=N双键(1.25 Å)和N—N单键(1.45 Å)之间。图5(c)显示68.3 GPa下γ-MgN8结构的三维电子局域函数。由图5(c)可知,N原子之间形成链状结构,每个N原子与相邻的两个N原子之间均有电子局域,形成共价键,键长为1.282、1.297和1.319 Å,3种N-N键的键长介于N=N双键(1.25 Å)和N—N单键(1.45 Å)之间。比较上述3相的N原子键长数据可以发现,β相和γ相的N-N键更容易断裂,释放出大量的能量,从而成为潜在的高能量密度材料。
图5(d)、图5(e)、图5(f)为MgN8晶体3种相ELF的二维切面图,晶格切面分别选取(010)、(110)和(010),ELF的数值范围为0~1。ELF = 1表示电子完全局域化,ELF = 0表示电子完全离域化或该处没有电子。从图5(d)、图5(e)、图5(f)中可以看出:在Mg原子与N原子之间存在电子局域,并且N原子附近对应的ELF值接近1,为高度局域的电子分布,说明Mg原子与N原子之间存在极性共价键;N原子与N原子之间对应的ELF值接近1,表明N原子与N原子之间存在共价键。
为了清晰地描述N原子与Mg原子之间的电子转移情况,计算了Bader电荷转移[23-24],计算结果见表3。N原子的电负性强于Mg原子,故N原子吸引电子的能力更强,Mg原子与N原子之间电荷转移的施主为Mg原子,受主为N原子。当压强为0 GPa时,α-MgN8相每个Mg原子向周围N原子的电荷转移为1.74e。由于该结构中的N原子不仅与Mg原子相连,还形成了N2和N3结构,N2与N3结构的存在导致N原子既可以作为受主,接收来自Mg原子和其他N原子的电荷,又可以作为施主,将电荷转移给其他N原子;当压强为24.3 GPa时,β-MgN8相中每个Mg原子向周围N原子的电荷转移为1.94e;当压强为68.3 GPa时,γ-MgN8相中每个Mg原子向周围N原子的电荷转移为2.20e。由此可见,随着压强的增加,电荷转移逐渐增多。
2.1. 晶体结构预测
2.2. 预测的MgN8晶体结构的稳定性
2.3. 预测的MgN8晶体结构的电子性质
-
运用CALYPASO软件并结合密度泛函理论第一性原理计算方法,对MgN8的晶体结构进行预测,同时对预测得出的晶体结构进行了结构优化。研究表明:在0~100 GPa范围内,3个相的相变序列为P4/mbm→P4/mnc→Cmcm;两次相变过程中,体积均发生坍塌,坍塌率分别为18.87%和2.62%。电子性质研究表明:α-MgN8相结构具有非金属性,带隙为3.093 eV;β-MgN8和γ-MgN8相结构具有明显的金属性。电子局域函数显示,Mg原子与N原子之间形成极性共价键,N原子与N原子之间形成共价键。高压β-MgN8和γ-MgN8相更适合用作储能材料,有望成为高能量密度材料的候选。Bader电荷转移计算结果表明,N原子具有更强的电负性,Mg原子与N原子之间电荷转移的施主为Mg原子,受主为N原子。在α-MgN8、β-MgN8、γ-MgN8相结构中,每个Mg原子向周围N原子的电荷转移分别为1.74e、1.94e和2.20e,即随着压强的增加,电荷转移增多。研究结果可以为碱土金属氮化物的进一步研究提供参考。