
 首页
首页 登录
登录 注册
注册




 下载:
下载:

全文HTML
-
氢是宇宙中含量最丰富的元素,拥有最简单的电子结构。然而极端压力条件可使常温常压下绝缘的氢经历一系列相变,最终可能演化为金属[1]。由于氢的原子轻,金属氢一方面被预测为室温超导体(德拜温度高,电声耦合强)[2],另一方面又被预测为超流体(量子效应显著)[3]。为验证这些预测,高压实验科学家付出了不懈努力以推高压力极限,不断逼近金属氢产生的实验条件。近年来,有一些报道宣称获得了金属氢[4-7],但因实验细节[8-12]存在争议且未获得重复实验,因此并未得到普遍认可[13-14]。在提升压力极限的过程中,表征金刚石对顶砧(DAC)中高压氢的技术手段也在升级,使得微小的氢样品可在200 GPa以上压力下通过微聚焦拉曼光谱[5,15-24]、可见光吸收光谱[18,25]、红外吸收光谱[4,6,17,20,24, 26-28]、电阻测量[5,7]等方法进行检测。遗憾的是,这些方法虽然可以提供氢的分子振动及电子结构信息,但是难以直接提供晶体结构信息。目前已经被公认,固体氢在400 GPa压力下至少存在5个物相[22]。研究氢在压力下晶体结构的演变,一方面对于理解氢的金属化过程至关重要,另一方面可以为理论计算中如何处理量子效应以及多体效应等问题提供有力的参考。但由于氢的X射线散射能力是所有元素中最弱的,想要通过X射线衍射方法直接测量高压氢的晶体结构绝非易事,而基于中子衍射的高压实验又难以达到足够的压力,导致氢的晶体结构研究滞后。长期以来,只有氢的第Ⅰ相晶体结构被明确确定为六方密堆积结构,而其他大多数高压相的晶体结构研究主要通过比对理论计算结果和光谱数据间接进行。
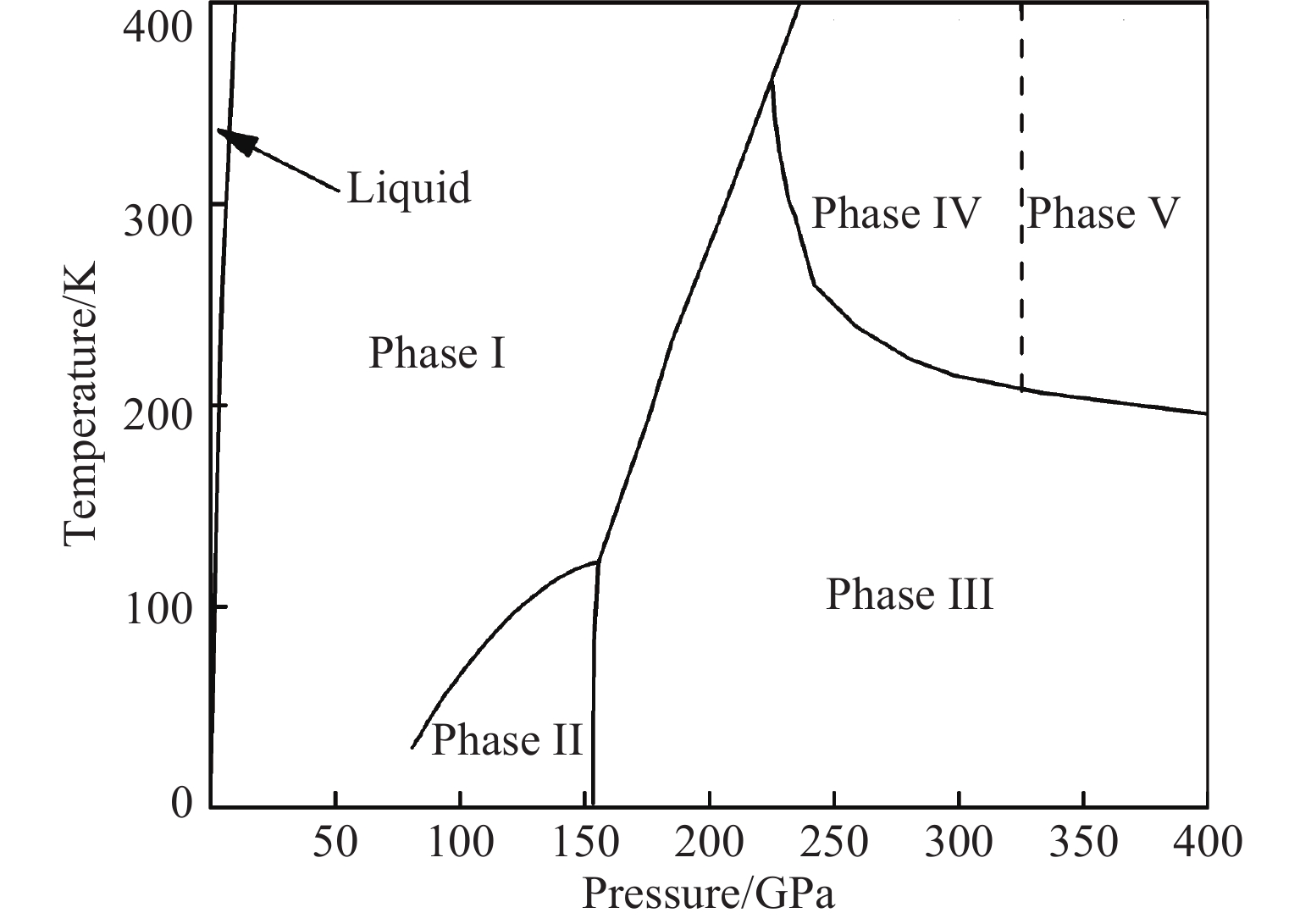
这里将对氢已知高压相的认识进行简要介绍。图1展示了氢的高压相图中描述固体相的区域[5,22,29-30],更高温度下发生的液-液相变可参见文献[31-34]。图1中虚线表示相边界是在室温条件下探测的,不同工作中的相边界温压条件存在不一致性。氢的第Ⅰ相具有六方密堆积结构。氢分子作为量子转子,其时间平均的电荷空间分布为球形(而非哑铃形)[35]。在低温下,氢(氘)在110 GPa[36](28 GPa[37])进入第Ⅱ相,氢分子由于压力不断增强的四极-四极相互作用由自由转子转变为“量子有序”,即分子的转动具有取向性。第Ⅱ相的相变条件受自旋异构和同位素效应的影响强烈[35],其他相则未明显表现出此现象。在低温条件下继续将压力升高至150 GPa以上[15],氢进入第Ⅲ相,分子停止转动进而成为“经典有序”。在室温条件下加压,氢在190 GPa进入第Ⅲ相,并在220 GPa以上进入第Ⅳ相[18]。第Ⅳ相中出现两个独立的振子,该特征与氢第Ⅰ、Ⅱ和Ⅲ相的振动光谱显著不同。继续升压,氢在325 GPa(第Ⅴ相)[22]再次经历相变,其光谱特征类似第Ⅳ相,也展现出两个独立振子。在室温条件、200 GPa以上氢展现出较密集的相变,且光谱特征新颖,因此我们将实验目标条件确定为室温和200 GPa以上。在理论计算方面,早期晶体结构分析主要是在立方密堆积结构的基础上研究氢分子取向有序造成的对称性变化[38-42]。将基于密度泛函的随机结构搜索应用于氢的晶体结构计算[43],使氢不同相的晶体结构涌现了更多候选,并趋于收敛[44-46]。理论计算预测第Ⅳ相中存在类石墨烯的类原子层和双原子分子氢层[44-45]可解释两个振子的现象。目前基于密度泛函得出的第Ⅲ相和第Ⅳ相结构模型的典型代表分别是C2/c[43]和Pc[44](或Cc[45])模型。然而对高密度氢进行理论计算仍然存在挑战,特别是如何更好地处理量子效应和多体效应等问题需近一步研究,譬如在密度泛函中的交换关联泛函[47-49]、路径积分分子动力学方法[50-51]、量子蒙特卡罗法[52-54]等方面,而最终理论方法的完善仍需要直接的晶体结构数据提供检验标准。
高压实验学家为推动固态氢晶体学的发展付出了不懈努力。美国华盛顿卡耐基研究院的Hazen等[55]最早对高压氢晶体结构进行测量。他们在接近氢固化的压力条件下(5.5 GPa[56])利用DAC制成晶体质量很高且样品尺寸较大的氢单晶样品,利用这些样品,即使在氢原子X射线散射截面非常小的情况下,也可通过实验室X射线单晶衍射仪获取氢的单晶X射线衍射数据[55]。然而,进一步升高压力会造成单晶破碎,导致X射线衍射信号迅速减弱,这就要求有更强的X射线源。毛河光等[57]在美国布鲁克海文国家实验室(BNL)的国家同步辐射光源(NSLS)搭建了可用来对DAC样品进行单晶X射线衍射信号采集的实验装置(使用白光和能量色散点探测器)。通过运用亮度提高数个数量级的X射线源,可在更高压力下测量氢的X射线衍射数据,最高压力可达到26.5 GPa。然而,即使使用同步辐射光源,在压力继续提升、晶粒破碎更加严重的情况下,之前的方法已无法延续。Loubeyre等[58]利用氢和氦的固体不互融原理在氦环境中生长氢单晶,使得氢单晶在高达百万大气压的条件下仍然能够保持足够大的晶体质量,从而利用毛河光等开发的单晶衍射方法在亮度更强的第三代同步辐射光源—欧洲同步辐射光源(ESRF)测量氢的X射线衍射结构数据,最高可测压力达到120 GPa。文献中记载的这个于1996年创下的室温压力记录保持了逾20年。随着大型面探测器的发展和成熟,基于单色光和面探测器的角散X射线衍射技术也被运用到氢的晶体结构研究中。Akahama等[59]在100 K低温条件下运用角散X射线衍射技术在日本SPring-8光源获得了氢的状态方程,最高压力达到180 GPa。该小组使用纯氢样品采集粉末衍射数据,得到十分微弱但仍可辨认的X射线衍射信号。近日,Loubeyre等[4]也报道了低温下190 GPa的X射线衍射数据,但该文献中没有介绍实验细节。然而,如欲在200 GPa以上测量氢的晶体结构,在获取压力和取得有效X射线衍射数据上的难度都显著增加,因此仍是一项困难的任务。
固态氘的高压中子衍射实验虽在氢的晶体结构解析上具有独特优势,但其发展滞后于X射线衍射实验。Goncharenko等[60]运用中子衍射(38 GPa)结合同步辐射X射线衍射(60 GPa),在低温下对氘的第Ⅱ相(X射线衍射至60 GPa)进行研究,发现了由氢分子取向的局部有序造成的对称破缺形成的非公度结构,这是目前利用中子衍射技术研究氢的压力记录。相较于X射线衍射,中子衍射对于研究氢的晶体结构有两个优势:一方面,氘的中子散射截面可观;另一方面,由于中子能够探测氘的原子核,对氘分子内原子核的分布和取向敏感,有利于探测氢高压相中分子取向有序性的信息。但由于中子束难以聚焦,而DAC样品获得高压的同时需要牺牲样品体积,因此在超高压下进行中子衍射仍然困难。近期发展的新型大腔体DAC在散裂中子源上的应用,可将高压中子衍射的压力提高至近100 GPa[61],但距离在200 GPa以上测量氢晶体结构仍然有相当大的差距,因此现阶段这个目标仍需要依靠X射线衍射的方法来实现。
我们投入近5年时间系统研究了在超高压下利用同步辐射X射线衍射测量氢晶体结构的方法,在室温条件下获得了高达254 GPa氢的X射线衍射数据[62]。在此工作中,我们研究了氢在等温(室温)压缩时从第Ⅰ相进入第Ⅲ相及第Ⅳ相的晶体结构变化,发现氢在第Ⅲ相和第Ⅳ相仍然具有类六方密堆积的晶体结构[62]。考虑到晶胞c/a值以及拉曼光谱的显著变化并结合理论计算,我们提出氢很可能经历了等结构拓扑电子相变[62]。对于早期工作中的科学发现[62]本文将不再赘述,以下将着重介绍和探讨实验工作的思路和方法。
-
欲在200 GPa以上压力通过X射线衍射测量氢的晶体结构须克服两大挑战:一是获得适合进行X射线衍射的超高压氢样品;二是对样品进行有效的X射线衍射测量。
通过恰当的样品制备方法获得样品是第一步。众所周知,氢的分子尺寸很小,在高压下极易钻进金刚石压砧表面的微裂纹,从而造成金刚石破裂,导致实验失败。因此,氢的超高压实验非常具有挑战性,一个成功实验的代价往往是耗费数十对金刚石压砧。为了减少氢渗透对金刚石的损害,主要可考虑3种解决方法:(1)利用低温条件降低氢的渗透性,然而,我们研究的目标物相是室温相,因此低温法无法采纳。(2)在金刚石表面镀薄膜阻止氢的渗透,该方法在氢的电阻测量[7]和红外光谱测量[6]中被报道过。然而,在200 GPa条件下,氢的样品厚度接近1 μm,而镀层厚度通常在几十纳米,考虑到X射线衍射强度和样品的电子数平方成正比,几十纳米厚的镀层也可能产生比氢信号更强的衍射信号,从而对辨认氢的信号造成干扰,因此该法也未被采纳。(3)通过缩小样品尺寸降低氢与金刚石砧面的接触面积,从而降低金刚石表面微裂纹暴露于氢的机会,该法主要用于拉曼光谱研究中。在拉曼光谱测量时,包裹氢样品的金属封垫相当于一个小光圈,不会对拉曼背底造成显著影响,而在X射线衍射实验中情况却截然不同,小样品周围的重金属封垫会产生强烈的X射线衍射背景信号,如图2(a)所示。图2中插图是样品的显微镜照片,红色虚线圈表示压砧的倒角面,绿色虚线圈内是氧化镁与环氧树脂的混合物。图2(a)和图2(b)原始数据中蓝框内为金刚石的衍射峰。图2(a)中的衍射环均来自铼和氢化铼。两个样品均使用30 μm以8.5°倒角至300 μm的金刚石压砧。两组数据均使用半峰宽为300 nm的聚焦光束,曝光时间均为60 s。根据小样品避免渗透的原则,我们通过改进样品制备方法(复合垫片)解决了这个问题,下文会对具体方法进行介绍。
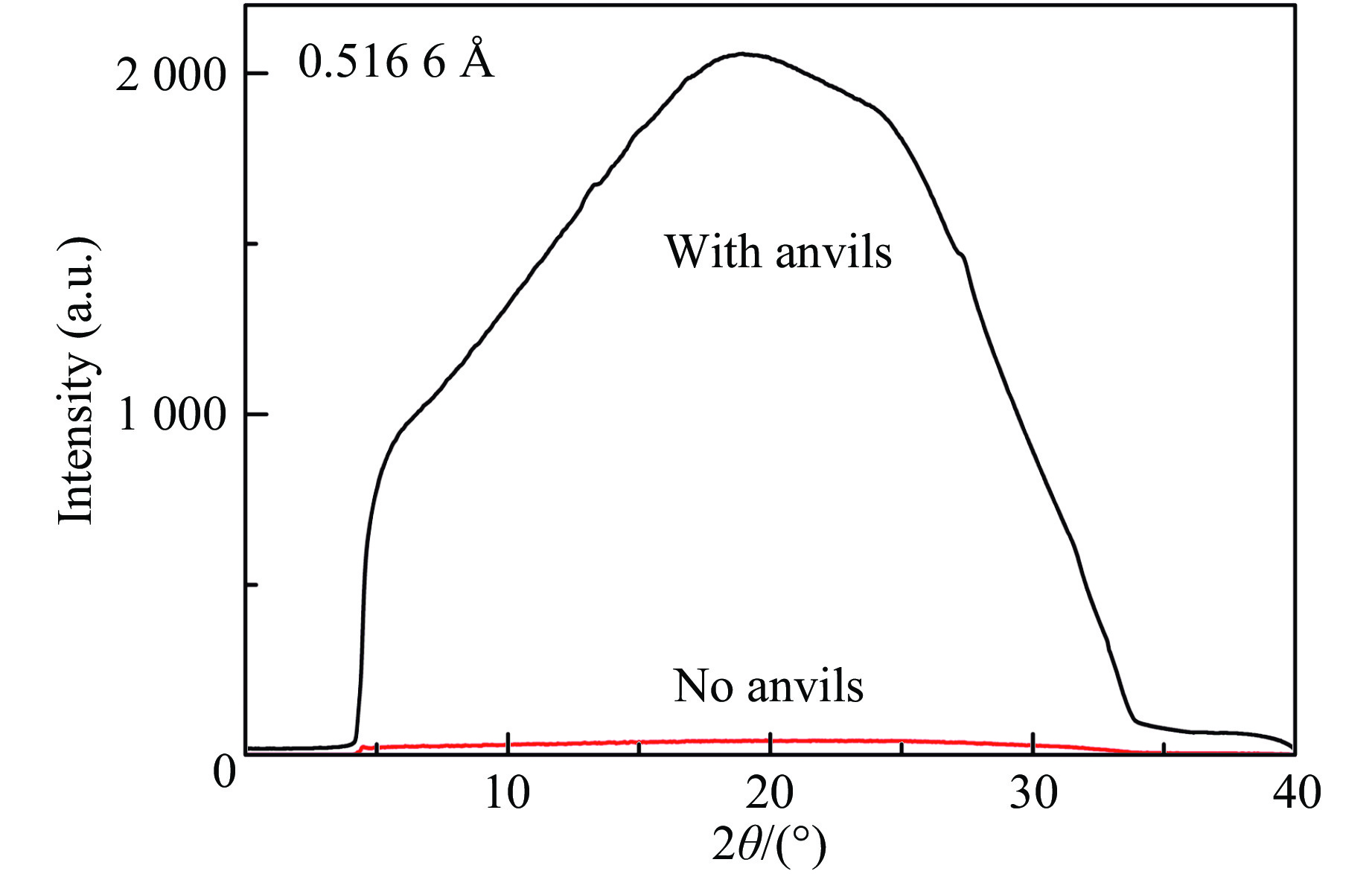
在获得超高压样品的基础上,更加困难的是获取氢的X射线衍射信号。为了获取200 GPa以上压力,使用砧面直径为20~50 μm的金刚石压砧,在200 GPa压力下样品体积为0.02 pL左右。超高压条件会导致晶粒破碎严重,这对于X射线衍射测量无异于雪上加霜。虽然传统金属封垫造成的衍射背底问题被复合垫片解决,但金刚石压砧康普顿散射造成的背底问题仍不可小觑,如图3所示,图中给出了同样曝光时间条件下有金刚石对顶砧(总厚度4.8 mm)和无金刚石对顶砧时的信号。经过探索,我们最后利用纳米光束X射线衍射和多通道准直器(MCC)等技术予以解决。
-
复合封垫技术完美解决了小样品金属封垫X射线衍射背底过强这一棘手问题,该问题也是超高压下进行X射线衍射实验的普遍难题。制作小尺寸样品是减少氢渗透得到超高压氢的一个关键举措。使用复合封垫制作小样品一方面提高了高压实验的成功率,另一方面为产生干净的X射线衍射谱提供条件。使用复合封垫时,氢样品周围近几十微米区域内没有重金属材料,取而代之的是衍射能力较弱的立方氮化硼或氧化镁与环氧树脂混合物,这样的几何构造会让入射聚焦X射线束的射线尾不直接轰击金属材料,从而使X射线衍射的背底变得干净。由此得到的氢样品在200 GPa以上压力下的直径通常为3~5 μm,仍然能够在X射线衍射实验中获得干净的背底,如图2(b)所示。这种复合封垫过去被应用于电输运测量中,插入复合材料的目的是使复合材料具有绝缘效果。此前,百万大气压以上条件下用复合封垫封装氢的实验只被Eremets小组报道过[7,63],我们的实验结果表明,此类复合封垫显著优化了超高压条件下X射线衍射数据的质量,为氢以及低原子序数材料的相关研究提供了重要帮助。
-
我们探索了Loubeyre等[58]提出的氦环境长氢单晶的方法,结果表明此法具有一定的可行性,但不适用于室温条件下、200 GPa以上压力氢的X射线衍射测量。在超高压条件下获得尺寸相对可观的氢单晶,主要考虑两种方法:一种是Loubeyre等提出的氦环境中长氢单晶的方法(该样品下文简称为氦氢样品);第二种是在超高压下通过反复熔化和凝固获取重结晶的大块氢单晶,该方法虽相当于在低压下[56]通过反复的加压、卸压(类似于降温升温)获取高质量的氢单晶,但要在百万大气压下通过加热获得大块氢单晶样品并不容易。最近,Zha等[23]发表的氢的熔化曲线显示,在100~300 GPa之间,氢的熔点虽从790 K下降至600 K,但仍远高于室温。对于将室温下就极具渗透性的氢加热到300°C以上以获取氢单晶,其制备难度远超出氦氢样品的制备方法,因此我们选择氦氢长氢法,在200 GPa以上的高压条件下,使用50 μm的金刚石砧面在直径15 μm左右的腔体内于氦环境中长氢单晶样品。经过将氢体积分数为20%的氢氦混合物加压至5.7 GPa(压力稍高于纯氢结晶压力[56]),氢开始结晶,通过精准控制加热和冷却,可在腔体中央长出一块高质量的氢单晶。通过30余次实验制得10个左右高质量样品,该样品可在各自最终能够达到的最大压力范围内保持相对良好的晶体质量,用来进行氢单晶的X射线衍射实验。利用波荡器插入件发出的高能单色X射线(20 ~37 keV)微米聚焦光束(典型尺寸为6 μm × 7 μm)结合面探测器(MarCCD165或Pilatus 1M)对样品沿Ω轴进行步进转动,采集单晶数据。通过使用Boehler-Almax设计的托块,在保证托块对金刚石侧向支撑的情况下,X射线开角可以达到40°(4θ)。实验结果表明:1 s曝光时间即可获取158 GPa下氢的X射线衍射数据[64]。单从信号强度的角度衡量,该方法前景乐观。然而,该方法仍旧面临两个难以逾越的障碍。首先,反复熔化凝固制成的氢单晶样品往往在超高压下形成强烈的择优取向,使得六方密堆积晶胞c轴倾向于沿金刚石对顶砧的压缩轴方向排布[64],测量到的氢衍射峰均为来自ab面内的峰,无法反映c轴的参数。若要克服障碍,需要加大压机的X射线开角。通过基于某样品实际数据的计算,开角需要扩大到60°(4θ)。增大开角意味着Boehler-Almax托块对金刚石支撑能力下降,可能会降低极限压力,因而未被采纳。其次,更大的问题在于氦氢样品难以获得更高压力,主要受两个因素限制。一是,氦原子的尺寸小于氢分子,作为传压介质的氦可能具备更强的渗透性,更易造成金刚石发生脆裂,导致实验失败。第二,由于长氢单晶的需要,样品腔的尺寸不能过小(通常直径在15 μm以上),使得强渗透性的氦与金刚石的接触面积过大,进一步导致压力难以提升。从欧洲同步辐射光源(ESRF)公开的实验报告中也可注意到Loubeyre等在发表了120 GPa的数据之后,曾尝试在室温下将氦氢样品做到更高的压力,但没有报道过显著提高压力的案例,随后Loubeyre等转向在低温下进行此类实验(因低温利于减少渗透造成的金刚石失效)。在超高压下维持可观尺寸氢单晶的努力受挫,促使我们调整思路,寻求测量破碎氢晶粒X射线衍射信号的方法。
-
我们的实验证明利用纳米聚焦光束可有效测量超高压下亚微米氢晶粒的X射线衍射信号,该方法与长氦氢样品的原理实质相同。氦氢方法的目标是维持一个尺寸和聚焦光斑(微米级光斑)匹配的结晶程度较高的氢单晶,而纳米衍射的目标是将X射线光子聚焦到一个亚微米晶粒当中,从而最大化X射线衍射的信号强度。纳米衍射方法的优点是允许使用很小的纯氢样品,从而有利于达到更高的压力。纳米光束衍射此前在对较高原子序数材料的高压研究中被成功运用[65-66],我们发现它同样适用于氢的超高压研究。
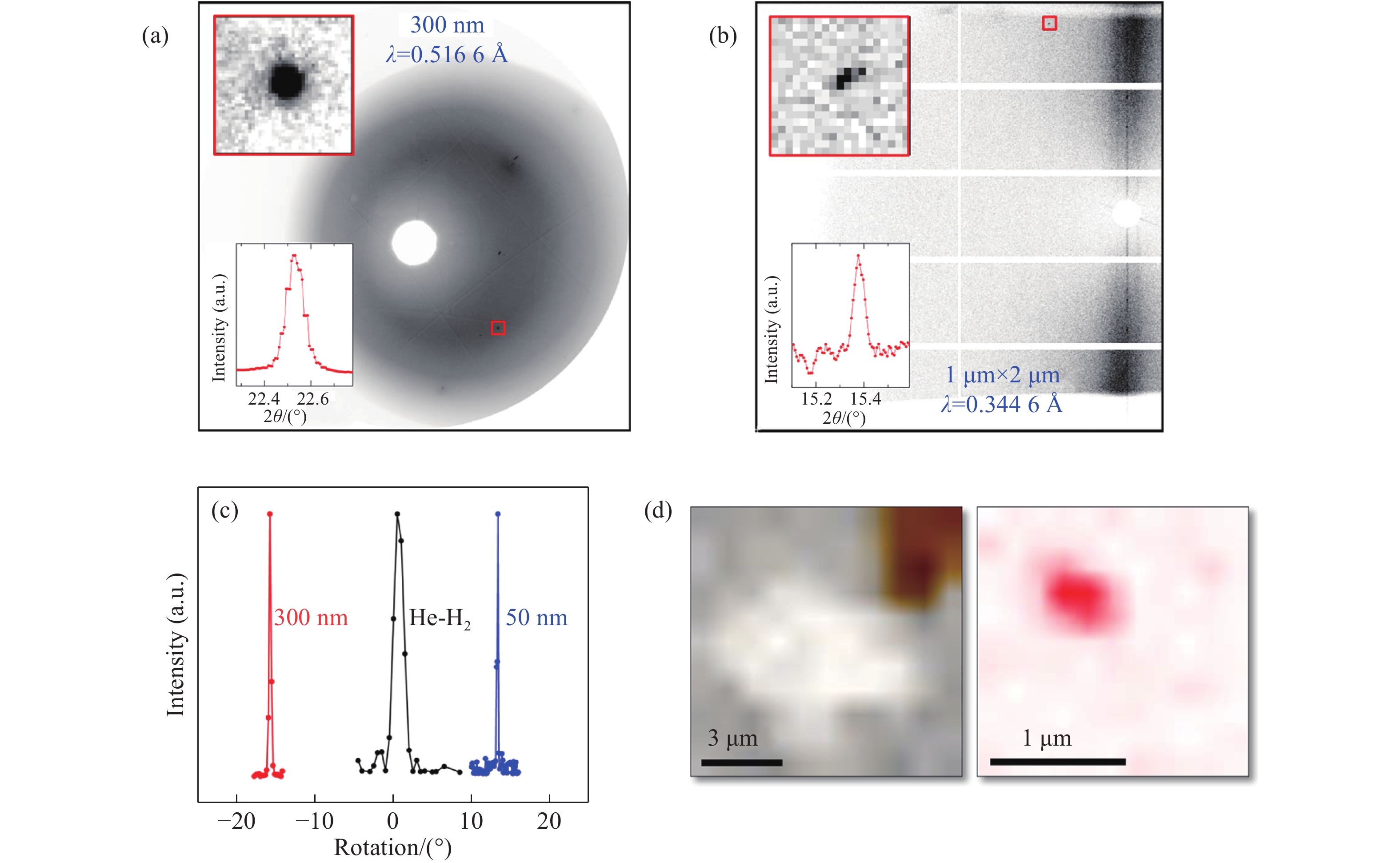
使用了两种光斑尺寸的纳米聚焦光束展开研究。在先进光子源的34 IDE线站,利用半峰宽为300 nm的单色光X射线束(24 keV)对样品进行转角法单晶X射线衍射数据采集(与氦氢样品的数据采集方法一致),得到了十分锐利的氢衍射峰。如图4(a)所示,红色方框内为氢的衍射峰,上插图为红框内数据的放大图,下插图为积分后的衍射峰。与氦氢样品的实验数据相比,利用纳米光束得到的数据中衍射峰多为斑点状,且信号背景比(信背比)提高了数倍。同时,数据显示晶体的镶嵌度较低,在232 GPa进行单晶数据采集时,摇摆曲线的半峰宽为0.4°左右,相比之下158 GPa测量氦氢样品的摇摆曲线半峰宽在2°左右,如图4(c)所示。进一步测量氢晶粒的尺寸,通过在与X射线垂直的二维平面中扫描样品并追踪选定的衍射峰,可绘制衍射晶体在二维平面上的投影。如图4(d)所示,在160 GPa压力下扫描了数十个晶粒,晶粒尺寸被确定为亚微米级且大于所使用的光斑尺寸(300 nm)。通过300 nm的X光束,准确测量了氢在254 GPa的X射线衍射数据[62]。目前,限制实验的主要因素是获取更高的压力(4.1节中将详细阐述)而非X射线衍射数据的质量。我们还试验了更小的纳米聚焦光斑,MAXIV作为最新一代的同步辐射光源(衍射极限环),其NanoMax线站可提供半峰宽为50 nm的聚焦单色X射线束(20 keV)。在200和220 GPa压力下的实验表明,50 nm光斑能够略微提高数据的信号背景比,测量的数据反映出更低的镶嵌度(摇摆曲线的半峰宽约为0.2°,较300 nm光束降低了一半)。以上结果表明,当使用的光束尺寸小于晶粒尺寸时,能够有效对氢样品进行X射线衍射测量。实验数据显示,在百万大气压以上,氢晶粒破碎为亚微米尺寸,但在160和200 GPa压力下的扫描数据显示晶粒并未随着压力升高发生进一步显著破碎,因而纳米聚焦光束有望在更高压力下对氢进行X射线衍射测量。最近,Loubeyre等[4]报道了利用红外吸收光谱在425 GPa、100 K条件下探测到可能的氢的金属化。根据第一性原理计算,该变化为C2/c-24结构转变到金属化的Cmca-12结构。纳米光束X射线衍射有望直接检验该温度和压力区间氢是否存在Loubeyre等提出的一级结构相变。
目前,使用纳米光束进行的X射线衍射实验技术存在一个缺陷。由于光斑和晶粒尺寸都很小,难以保证在转动样品进行数据采集的过程中X射线能够锁定其中一颗亚微米晶粒,使得采集到的数据为在一定样品范围内来自不同晶粒的统计结果,这些数据对于填补氢在超高压下晶体结构数据的空白有重要意义。若要进一步对氢的晶体结构模型进行精修,则需要对现有技术进行升级,例如提高样品转台及平移台的精度和稳定性,提高棚屋内的温度稳定性,优化聚焦光斑的尺寸以覆盖整个亚微米晶粒等,从而获取来自同一颗亚微米晶粒产生的X射线衍射数据。
-
MCC作为微区准直装置,可以采集来自微小样品的信号并有效屏蔽周围背景,我们将其成功应用于200 GPa以上氢的X射线衍射研究中。在高压实验中,MCC早期被用于同步辐射大体积腔体压机的X射线相关实验[67-68],且多用来采集高压液体的微弱散射信号。近年来,MCC的应用扩展至DAC实验中,成功测量了低压下的液态氢结构[69]以及百万大气压下的SiO2玻璃[70]结构。早期为研究液体散射设计的MCC存在垂直方向X射线散射角过小(10°)的缺陷,十分不利于单晶衍射实验,主要由于液体散射和单晶衍射存在显著区别。液体散射信号通常是沿着X射线轴向对称分布,即使是10°的小开口也可采集到可靠的液体散射信号,但单晶衍射峰很可能出现在这个10°窗口范围之外的位置。为了更好地采集超高压氢单晶的XRD数据,美国阿贡国家实验室先进光子源高压研究团队(HPCAT)开发了新型增大垂直开角(30°)的MCC,这种MCC结合大面积二维探测器(Pilatus 1M),X射线出射信号基本可以覆盖整个探测器。利用该MCC结合微米聚焦光束(半峰宽为2 μm × 1 μm),可在250 GPa压力下采集到氢的X射线衍射信号,如图4(b)所示。我们的实验表明,MCC可以使来自金刚石的康普顿散射信号大幅减弱,信背比提高近5倍。值得一提的是,金刚石的康普顿散射信号是DAC实验的主要背景信号(图3),尤其是在测量超高压实验中样品量非常少而且样品为低原子序数材料时(样品本身信号很弱,降低背景信号强度至关重要)。
最后需要指出使用MCC会带来两个副作用。首先,MCC在几何上是由两层平行放置的金属薄片形成的狭缝,因而在过滤来自样品腔外信号的同时,也会阻挡来自样品的信号,导致信号的采集时间大幅增长。在我们的实验中,利用MCC采集一个XRD谱所用时间可长达20 min,而同样的入射光强测量非低原子序数材料单晶样品的曝光时间通常在1 s以下。在高亮度X射线下,曝光时间增长一方面让原本就机时资源紧张的同步辐射实验变得不经济,更重要的是会提高金刚石的损害风险(4.1节中阐述),从而令实验提前终止。其次,MCC被置于样品和探测器之间,增加了样品和探测器的距离,从而减小数据在倒易空间的覆盖范围,这对于能量较高的线站(30 keV以上)不会造成太大影响,但是对于在20 keV左右能量操作的线站,就要求使用更大的面探测器进行弥补,或是采用多个探测器位置进行数据采集(成倍数增加曝光时间)。但总体而言,大开角MCC的开发为超高压下氢以及其他低原子序数材料的晶体结构研究提供了一种有力工具。
3.1. 复合封垫技术
3.2. 氦环境下氢单晶的生长方法
3.3. 纳米聚焦光束X射线衍射
3.4. 多通道准直器(MCC)
-
本节将讨论与实验和数据分析相关的3个问题,探讨这些问题对于成功开展实验和正确解读数据具有重要的意义。
-
在实验中发现,长时间的X射线辐照对于通过旋转法长时间曝光采集数据的超高压氢样品不利,易造成金刚石破损。得出这样的经验性判断是基于下述观察。从获得高压的角度,将新样品直接加压至特定高压再采集X射线衍射数据的成功率优于逐渐加压并测量获得数据。具体来说,前期实验中我们对氦氢样品采取逐渐加压,在每个压力点采集单晶衍射数据。这种做法一方面是为了丰富状态方程的数据点,另一方面为了方便追踪越来越弱的衍射峰。对于MCC测量的大多数纯氢样品(使用10°垂直开角MCC)也遵循了逐渐加压和测量的操作,以便在低压下先锁定两条氢的衍射峰,从而在高压下不需要大范围扫描转角进行搜索(MCC的曝光时间长且窗口太小)。这些步进加压并测量样品的压力最终都没能超越210 GPa。后期实验中,每个样品都在几分钟内被快速加压至目标压力,立即进行X射线衍射数据采集。原则上一个压力点对应一个样品(不同样品的数据可通过4.3节中的d100压标进行统一)。在该种操作下,得以在210~254 GPa压力范围内获得了氢的XRD数据。还需要强调的是,即使在每种压力下使用一个“新鲜”样品进行测量,大量的样品仍然会在数据采集过程中发生金刚石破裂。文献中报道的以及我们自己对其他样品的超高压实验中,并未观察到明显的由辐照造成的金刚石压砧破裂。本次实验相较于以往实验有3点显著不同:第一,样品是氢,具有高度渗透性;第二,本次实验需要对辐照中的样品做 ± 20°转动,由于转动会造成X射线穿过金刚石的体积大幅增加,大多数超高压实验测量粉末样品不需要在信号采集过程中对样品进行转动操作;第三,谱的采集时间长。在纳米聚焦光束的实验中,每个谱的采集时间为60~120 s;而使用MCC的实验中,采集时间通常在5~20 min。 相较之下,波荡器线站对高压下的高原子序数材料进行数据采集所需的曝光时间只要几秒。如果样品是单晶,曝光时间往往低于1 s。虽然由辐照导致的金刚石失效的具体机理不明,但是在对氢进行超高压XRD研究时,我们仍建议优化X射线的剂量,以尽可能小的入射剂量取得可以接受的X射线衍射信号,而非入射线束亮度越高越好。在某些高亮度的波荡器线站,甚至要考虑削减X射线的强度。另外纳米光束从减少辐照损伤的角度也占有优势:一方面能够减少与金刚石接触的体积;另一方面,纳米光束的通量密度高,但是总通量不高,有利于保护金刚石。
-
与普通单晶XRD数据不同,辨认超高压氢的衍射峰需要额外的努力。虽然利用纳米聚焦光束得到的衍射峰多为点状,但与一般材料的单晶衍射强度相比,强度普遍不高,信号强度(Is)与背景强度(Ib)的比值在1.2~10范围内[64]。同时由于数据采集时间较长,探测器上会出现多种来源的杂质信号。Dubrovinsky等[71]对我们工作[62]中衍射峰的选取提出了质疑,称我们没有对原始数据谱中出现的所有衍射点给予充分解释。实际上,所有被Dubrovinsky等标注的衍射点之前已经被逐一检查过,并排除了来自于氢的可能。我们主要通过4个标准判断探测器上的点是否来自于氢样品,即:重复性(同一d值的衍射峰多次出现)、峰形(衍射点多为圆形,少数由于晶体的应变变成短弧形)、峰宽(积分后一维曲线中的2θ峰宽)以及摇摆曲线的半峰宽。以文献[62]中232 GPa压力数据为例,该数据是使用300 nm聚焦光束通过MarCCD165探测器采集的。首先有40个衍射点被判定为来自于氢,其中有8个(100)、5个(002)以及27个(101)。这3类峰的d值方差/平均d值分别为0.000 9 Å/1.479 5 Å、0.001 Å/1.322 Å和0.000 9 Å /1.291 0 Å,可见每一族的衍射峰都能找到多个重复数据点。而且这些峰的峰形多为圆形,积分峰宽和摇摆曲线也都类似。积分峰宽(2θ)为0.1° ± 0.01°(波长0.516 6 Å),摇摆曲线的半峰宽为0.3° ± 0.1°。另外,由于曝光时间长,采用CCD探测器时会产生一定量的电子暗电流信号,这些电子噪音通常只占据非常少的像素点,因而可以较清晰地辨认。还存在一些积分峰宽非常宽的衍射点(2θ半峰宽通常大于0.2°),这些峰摇摆曲线的半峰宽通常也很大(可达数度)。这些峰是由存在很大应变的金刚石压砧或封垫材料产生,或是这些信号所产生的多次散射,剩余不存在重复性的疑似信号是由宇宙射线等偶发事件产生。通过了以上判据的实验数据才被认定为来自于氢的信号。
-
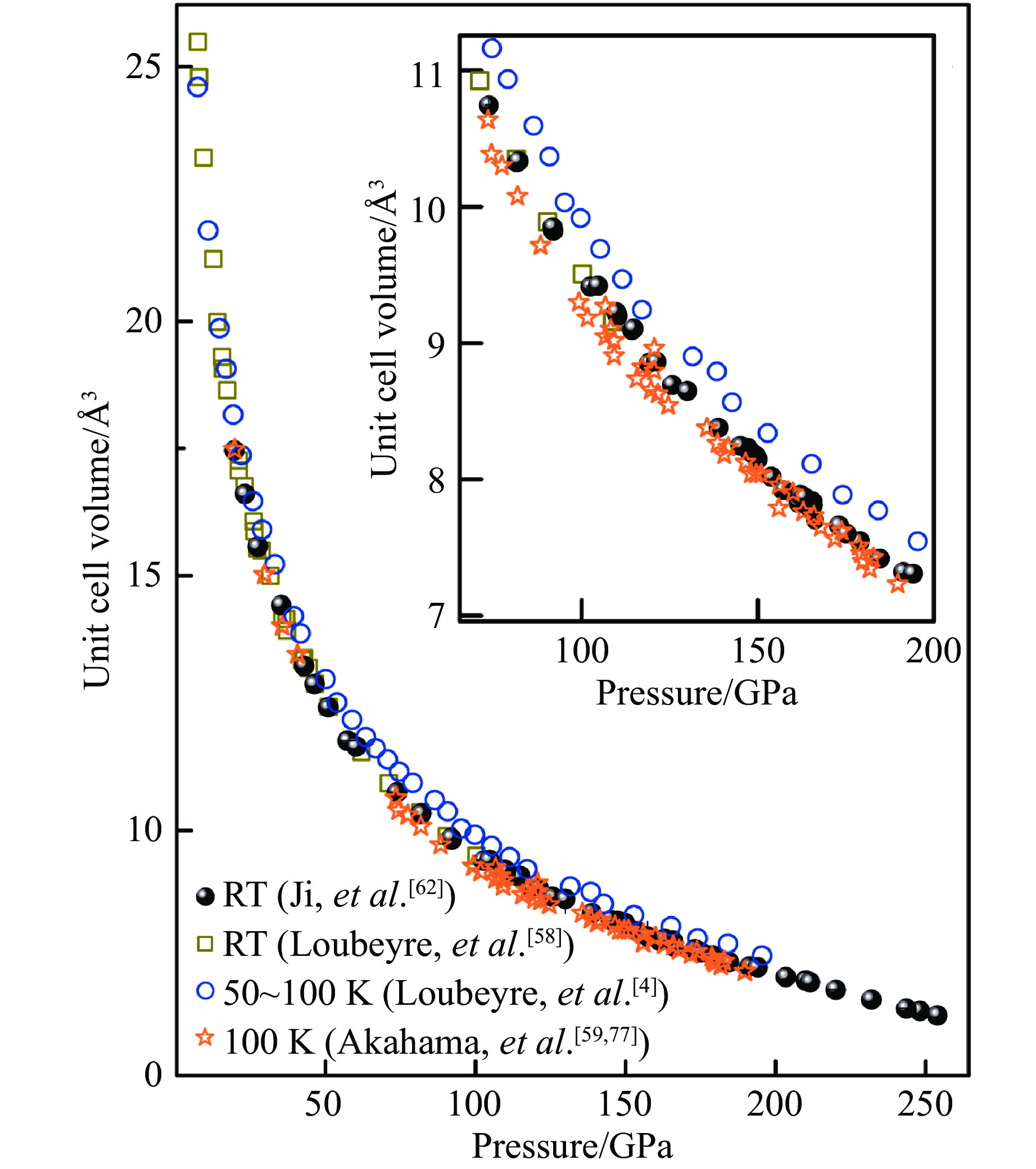
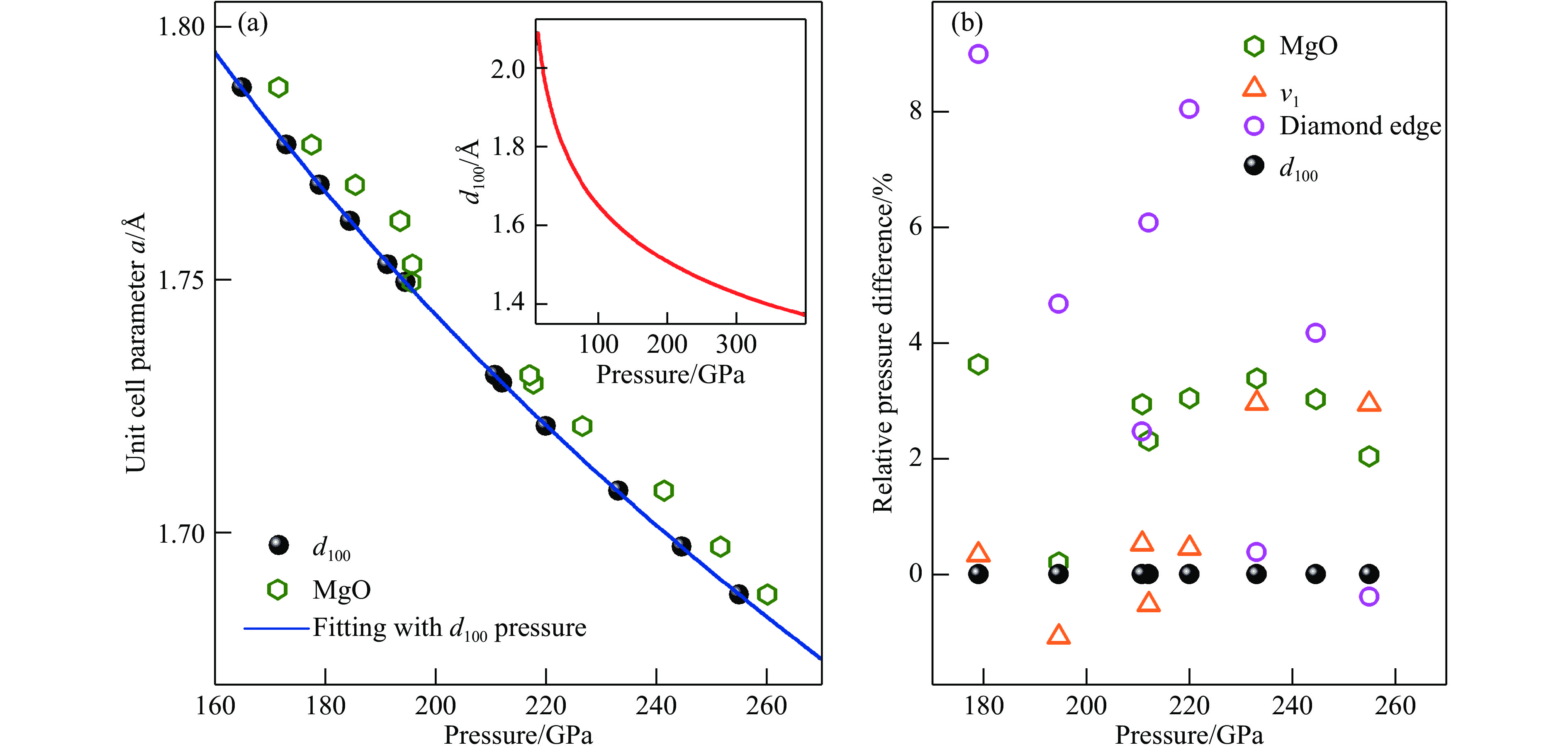
标定压力对于统一和对比不同数据氢的状态方程十分重要,我们提供了一种基于氢X射线衍射数据的压标。在超高压下准确测量压力是一件棘手的任务。目前,在超高压范围内缺乏一个获得广泛认可的单一压标。这个缺憾的后果十分明显,如图5所示,不同研究者的低温数据(Arkahama等[72]和Loubeyre等[4])在200 GPa以内标压的系统偏差可达到近10%。为弥补缺乏单一压标的缺憾,我们的实验中使用了多种压标来保证压力的可靠性,具体有红宝石荧光压标(60 GPa以下低压范围)、金的状态方程压标[73](最高压力至160 GPa)、氧化镁状态方程压标[74](以氧化镁为填充物的复合封垫样品)、氢的100族d值压标(简称d100压标)[62]、金刚石拉曼边压标[75]以及氢的振子压标[76]。其中d100压标是利用金的状态方程压标标定出来的。我们使用金压标在室温条件下将d100和压力的关系标定到158 GPa,并将压力-d100曲线外插至更高的压力作为压标使用。因为氢的压缩性很强,体积变化对压力十分敏感,且该压标直接反映了氢样品本身的压力,此压标可为后续氢的XRD实验提供重要参考。然而,从拉曼实验可知,氢在室温条件下于190和220 GPa分别经历两次相变[18],因此需要确定两次相变的边界d100随压力的变化是平滑的。我们将复合垫片中氧化镁填充物与氢接触部分的压力和由d100推算出的压力作了比较[64],如图6(a)所示,其中蓝色曲线是基于d100压标绘制的晶胞参数a-压力拟合曲线,插图给出了100~400 GPa范围的压力-d100曲线。d100在两种压标下随着压力的移动并未因为第Ⅰ相至第Ⅲ相以及随后至第Ⅳ相的相变而发生明显不连续变化,证明了将压力-d100曲线外推至高压具有合理性。Dubrovinsky等[71]对我们使用d100压标标定第Ⅲ、Ⅳ相压力提出了质疑,上述论述阐明了使用该压标的理由。另外,我们将不同压标在200 GPa以上的表现进行了横向对比,如图6(b)所示。定量来看,在200 GPa以上将其他压标与d100压标进行对比,氢的振子压标和d100压标最为接近,最大偏离3%;氧化镁压标系统性将压力高估3%~4%;金刚石拉曼边压标则最高偏离9%。由于d100与氢振子压标均为来自氢本身的压力,两者最为符合,也最为可靠。氧化镁压标反映的压力被系统性的高估,可根据高估比例进行修正。金刚石拉曼边压标则相对最不可靠,这与文献[76]相符,因为该压标标定的压力受到多方面因素影响,如金刚石压砧的形貌、样品的尺寸、封垫材料、封垫的几何尺寸等。在室温和更高压力下,据报道氢在325 GPa[22]压力下再次发生相变。光谱数据[18-19,22]显示第Ⅴ相在结构上变化不大[46],因此d100压标很可能仍适用。我们给出了该压标外推至400 GPa的数据[64],但后续仍需对该压标与其他压标进行横向标定,以保证压标在相变后的准确性。
4.1. 与X射线辐照相关的金刚石压砧破坏
4.2. 氢衍射峰的辨认
4.3. 基于氢X射线衍射的压标问题
-
通过系统性的研究,运用同步辐射纳米聚焦光束和MCC技术结合复合封垫技术,将氢的晶体结构测量压力提高至254 GPa[62]。从技术角度来看,这些实验方法适用于将氢的晶体结构研究延伸至更高压力。与此同时,仍有挑战亟待突破。譬如,如何有效地采集来自于一个孤立单晶的X射线衍射数据?如何采集到101族以上的弱衍射信号以提高数据在倒易空间的覆盖率?这两个问题决定了能否利用X射线衍射数据对氢的晶体结构模型进行精修。另外由于X射线衍射对于探测氢原子位置的先天不足,能够推广至更高压力的中子衍射技术同样亟待突破,以对X射线衍射数据进行必要的补充。总而言之,在同步辐射技术以及先进高压技术[65, 78-80]不断进步的背景下,在更高压力条件下对氢甚至是金属氢的晶体结构进行直接测量指日可待。