
 首页
首页 登录
登录 注册
注册



-
在过去的几十年里, 随着全球对清洁能源需求的增加, 太阳能电池作为一种重要的可再生能源受到了广泛关注. 目前, 开发用于太阳能收集的新材料是可再生能源领域的主要研究课题之一. 自第一代硅基太阳能电池问世以来, 研究人员陆续发现了二元硫族化合物碲化镉(CdTe)[1]、四元硫族化合物铜铟镓硒(CIGS)[2]和铜锌锡硫(CZTS)[3]以及钙钛矿(perovskite)[4]. 在这些新兴的吸收层材料中, 具有四面体配位结构的半导体材料一直是太阳能电池研究领域的热点之一[5]. 其中, 锌黄锡矿(kesterite)相的CZTS作为一种直接带隙半导体, 因其较高的光吸收系数、合适的带隙(1.39—1.52 eV)、元素含量丰富, 在低成本薄膜太阳能电池应用中展现出巨大的优势. 然而, 目前基于CZTS材料制备的太阳能电池最高光电转换效率(PCE)仅为14.9%[3], 远低于同为锌黄锡矿相的CIGS太阳能电池23.6%的PCE[6]. 这种差距主要归因于CZTS太阳能电池的开路电压损失(Voc deficit)为650 mV[7–9], 而CIGS太阳能电池的开路电压损失为500 mV[10]. 开路电压损失主要来源于材料内部和界面处的缺陷引起的非辐射复合. 理论研究表明[11–13], 在CZTS中, CuZn + ZnCu反位复合缺陷是主要缺陷, 由于Cu+和Zn2+离子的半径相似(均为0.74 Å), 并且Cu+和Zn2+离子具有相同的四面体配位环境, 因此CuZn反位缺陷具有较低的形成能, 导致材料容易产生CuZn反位缺陷. 而CuZn是一种深能级的受主缺陷, 因此会作为载流子的非辐射复合中心, 从而降低电池的开路电压. 此外, Sn4+的离子半径(约为0.69 Å)也与Cu+和Zn2+接近, 导致材料也容易形成2CuZn+SnZn的复合缺陷[11]. 这些深能级缺陷使得CZTS比CIGS太阳能电池表现出更多的非辐射复合. 另外, 在CZTS中的3种阳离子配位数相同, 容易形成大量的缺陷和缺陷簇, 导致带隙尾态较大[14], 也会导致电池效率的下降.
针对上述问题, 从改进CZTS的物理性质, 尤其是改善缺陷性质出发, 利用离子尺寸差异更大的同价态元素对CZTS中的元素进行部分替代, 是一种最直接且可行的研究方案. 例如, 用Ag+(半径约1.14 Å)部分替代Cu+, 可以显著减少CuZn反位缺陷, 同时还可以调节能带结构, 但随着Ag含量的上升, 材料的性质从p型转向n型, 这对CZTS太阳能电池不利[15–19]. 目前利用Ag掺杂的CZTS制备的电池PCE也只达到了12.5%[16]. 同时有研究表明, 将Cd2+(半径约0.92 Å)掺入CZTS后, 可以减少二次相ZnS的形成[20,21], 并且可以改善带隙尾态的问题[22]. 研究还发现, Cd2+的掺杂可以抑制CuZn和ZnCu的反位复合缺陷, 提高载流子浓度, 从而提高电池效率, 但随着Cd的含量增大, 材料从锌黄锡矿相转变为黄锡矿(stannite)相[23]. 目前以掺入Cd的CZTS制备的电池光电转换效率达到了12.6%[24]. 此外, 有研究人员从材料设计的角度出发, 采取同价态元素替代方案, 设计了新型四元硫族化合物材料, 并进一步研究了其热力学稳定性以及电子结构等性质. 例如, Wang等[25]通过第一性原理计算, 研究了锌黄锡矿相四元硫族化合物I2-II-IV-VI4 (I为Cu; II为Zn, Cd, Hg, Mg, Ca, Sr, Ba; IV为Ge, Sn, Ti, Zr, Hf; VI为O, S, Se, Te)的热力学稳定性. 研究结果表明当II为Zn, Cd, Hg; VI为S, Se, Te的锌黄锡矿相化合物是稳定的. 而当II为Mg, Ca, Sr, Ba; IV为Ti, Zr, Hf以及VI为O的锌黄锡矿相化合物是不稳定的, 容易分解成二元或三元的二次相(secondary phase). 但是在后续的工作[26–28]中, 研究人员成功合成了四元化合物Cu2BaSnS4和Cu2BaSnSe4. 研究表明, 由于碱土金属Ba2+离子半径较大(约为1.49 Å), Ba2+不再处于四面体配位中心, 因而会形成非锌黄锡矿相的结构. 同时, 第一性原理计算表明[29–31], 在Cu2BaSnS4(Cu2BaSnSe4)中, 由于Cu+和Sn4+仍然保留了四面体配位环境, 因此它们表现出类似于CZTS的电子结构和光学性质. 同时, 由于Ba2+的离子性, 相比CZTS, 它们对缺陷容忍度更高. 计算结果表明, 大多数与Ba相关的缺陷由于其较强的离子性表现出较浅的缺陷转变能级. 并且BaCu和CuBa反位缺陷的形成较高, 材料的主要缺陷与CIGS类似, 仍然是浅能级的Cu空位(VCu)缺陷. 因而该体系可以缓解阳离子无序和带隙尾态的问题. 目前以CBTS为吸收层制备的太阳能电池的效率已达到6.17%[32].
另外, 有研究表明利用异价元素对CZTS中的阳离子进行部分替代, 也可以有效改善材料的性能. 如Du等[33]将Ga3+掺如CZTSSe, 得到了光电转换效率12.3%的CZTS太阳能电池. 通过第一性原理计算研究[33], 发现Ga3+阳离子优先占据Zn2+和Sn4+的晶格位, 可以有效地抑制SnZn深能级受主缺陷. 并且, 掺入Ga3+后, 还可以形成如GaZn+ CuZn和GzZn+GaSn缺陷-掺杂剂团簇, 可以改善带隙尾态. Kuo和Tsega [34]将In3+掺入CZTSe, 制备了p型材料Cu1.75Zn(Sn1–xInx)Se4 (0 < x < 0.6), 实验表明随着In掺杂水平增加, 材料载流子浓度先增大后减小, 在x = 0.4时达到最大, 为8.0 cm2/(V·s), 表明In掺杂可以改善CZTSe的电学性能. 除此之外, 碱金属由于其较强的离子性, 普遍显现出较高的缺陷容忍度, 因此碱金属掺杂对CZTS性质的影响也逐渐引起了研究人员的关注. 如Sun等[35]采用Li+(约0.59 Å)和Na+(约0.99 Å)的共掺杂策略协同调控CZTS. 研究发现在掺入Li后, 有害的深能级CuZn反位缺陷的浓度降低了8.13%, 而Na掺杂后, 良性的NaZn反位缺陷浓度增大了36.91%. 因此, Li和Na的协同掺杂会显著提高载流子浓度, 并且界面缺陷浓度降低了一个数量级. Maeda等[36]通过对碱金属原子在锌黄锡矿相CZTS中的扩散机制的研究, 证实了碱金属元素除了可以同价替代Cu+以外, 还可以异价替代Zn2+. 研究还表明只有半径较小的Li+, Na+离子能进入晶格, 而K+离子由于其离子半径较大(约为1.37 Å), 难以替代Cu+或Zn2+, 退火时K会在晶界以K-Se二元化合物的形式析出.
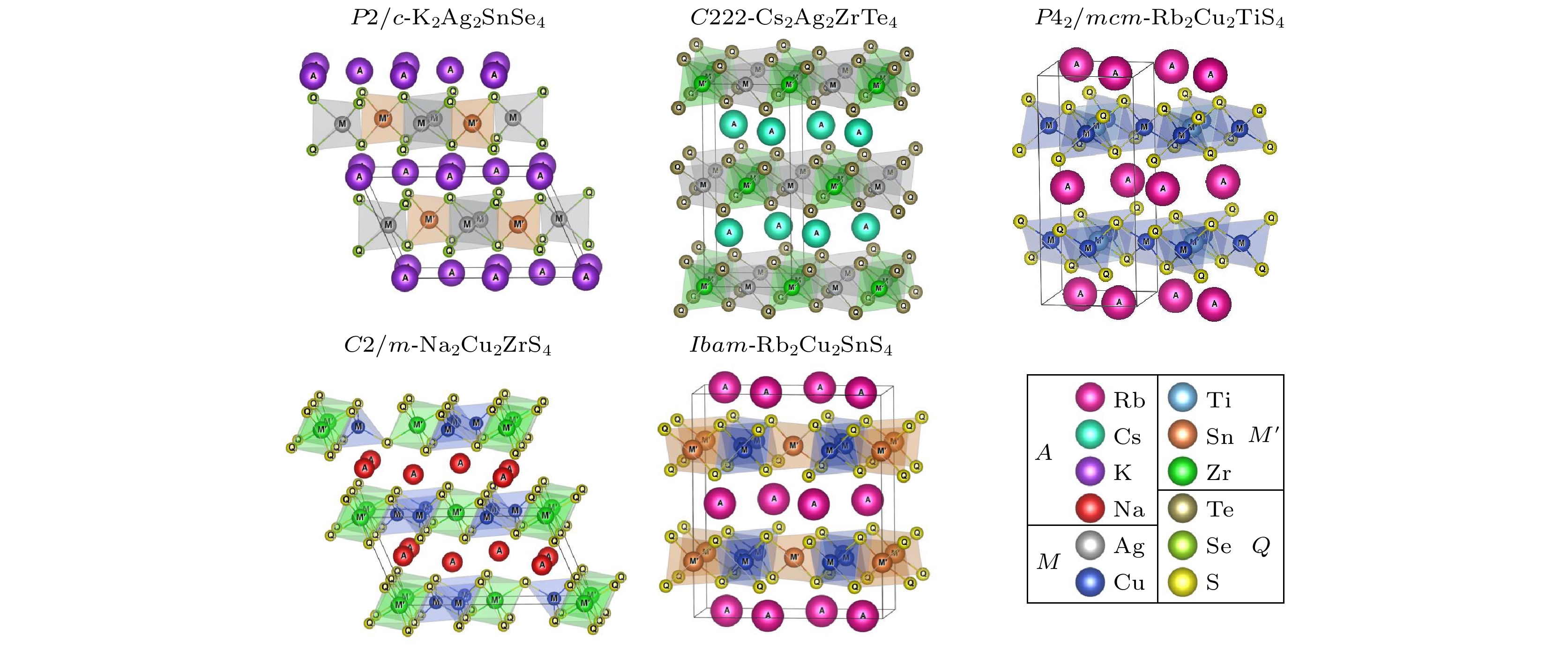
值得注意的是, 采用异价元素替代设计新型太阳能电池材料也是一种可行的策略. 从ZnS设计出CIGS和从CIGS中设计出CZTS材料[37], 是采用该设计策略最为成功的例子. 考虑到碱金属离子较高的缺陷容忍度, 利用碱金属元素对CZTS中的Zn进行异价替代, 从而设计新型光伏材料, 该设计策略的可行性与材料的物理性质均值得进一步研究. 实际上, 目前实验上已有非锌黄锡矿相的Rb2Cu2SnS4, Rb2Cu2TiS4和K2Ag2SnSe4等四元硫族化合物被成功地合成[38–46]. 其晶体结构如图1所示. 以Rb2Cu2SnS4为例, 这种材料可看作CZTS中一个Zn2+离子被两个Rb+碱金属阳离子替代所形成的新化合物. 这种化合物表现出层状结构, 其中一层为[CuS4]四面体与[SnS4]四面体共享边所构成的[Cu2SnS4]2–的负电层, 这是一种带缺陷的反氧化铅状的结构[46], 另一层则是Rb+碱金属阳离子层. 实验测得其带隙为2.04 eV, 可能适用于光伏发电和光电化学分解水应用[29]. 可以看出该类化合物A2M2M'Q4 (A = Na, K, Rb, Cs, In, Tl; M = Cu, Ag, Au; M' = Ti, Zr, Hf, Ge, Sn; Q = S, Se, Te)与CBTS类似, 大尺寸的碱金属阳离子会造成CZTS晶格产生较大的畸变, 因此这类材料均不会形成锌黄锡矿相. 同时该类材料还保留有四面体结构单元, 因此可能具有与CZTS类似的电子结构和光学性质[31]. 另外, 由于加入了离子性较强的碱金属, 这类材料可能会具有比CZTS更高的缺陷容忍度. 因此, 有必要对该类四元硫族化合物的电子结构、光学等特性进行更深入的研究.
在本工作中, 对目前已知的A2M2M'Q4四元硫族化合物的结构进行分析, 通过元素替换, 建立了一个包含1350种A2M2M'Q4的材料数据库. 并对新生成的材料进行了第一性原理高通量计算, 以寻找高效的太阳能电池吸收层材料. 通过计算材料的热力学稳定性、带隙、光谱极限最大效率(spectroscopic limited maximum efficiency, SLME)、声子散射谱等特性, 筛选出10种稳定的、理论光电转换效率超过30%的A2M2M'Q4化合物. 本工作的研究结果为未来A2M2M'Q4化合物在光伏领域的应用提供了理论基础.
-
本工作中, 筛选新光伏材料的高通量计算工作流程如图2所示. 该流程共分为3个部分. 首先为预筛选过程(pre-screen), 该流程主要目的是通过元素取代, 构建具有合适晶体结构的A2M2M'Q4材料数据库. 从Material Project数据库[47]中获取四元硫族化合物A2M2M'Q4的初始结构, 并筛选出含有四面体结构单元的晶体结构. Material Project数据库中获得到的A2M2M'Q4材料具有9种空间群, 它们分别是: Rb2Cu2SnS4[38](空间群: Ibam, #72), K2Au2SnS4[38](空间群:
${P}\bar{1} $ , #2), Cs2Au2SnS4[39](空间群: Fddd, #70), K2Ag2SnTe4[40] (空间群:$ {I}{4}2{m} $ , #121), K2Ag2SnSe4[41] (空间群: P2/c, #13), Rb2Ag2GeS4[42] (空间群: C2/c, #15), Cs2Ag2ZrTe4[44] (空间群: C222, #21), Na2Cu2ZrS4[43](空间群: C2/m, #12), K2Cu2TiS4[45] (空间群: P42/mcm, #132).如图1所示, P2/c-K2Ag2SnSe4, Ibam-Rb2Cu2SnS4, C222-Cs2Ag2ZrTe4, P42/mcm-K2Cu2TiS4和C2/m-Na2Cu2ZrS4这5种具有不同空间群的化合物中, 均存在[MQ4]或[M'Q4]四面体结构单元. 然后, 我们通过对这5种已合成的材料进行同价态元素替代生成新的四元化合物: A位点用碱金属元素(Na, K, Rb, Cs)和ⅢA族元素(In, Tl)进行替换; M位点用IB族元素(Cu, Ag, Au)进行替换; M' 位点用ⅣB族元素(Ti, Zr, Hf)和ⅣA族元素(Ge, Sn)进行替换; Q位点用ⅥA族元素(S, Se, Te)进行替换. 通过该流程我们生成了1350个A2M2M'Q4四元硫族化合物, 接下来将对生成的化合物进行第一性原理计算, 筛选出最具光伏应用前景的材料.
通过第一性原理计算了A2M2M'Q4硫族化合物的凸包上方能量(energy above convex hull, Ehull)和带隙(
$ {{E}}_{{{\mathrm{g}}}}^{\text{PBE}} $ 和$ {{E}}_{\text{g}}^{\text{HSE}} $ ). 筛选标准为热力学稳定(Ehull = 0)和亚稳定 (0 < Ehull < 0.05 eV/atom)的化合物, 以及PBE带隙$ {{E}}_{\text{g}}^{\text{PBE}} $ 范围在0.4—1.2 eV, 直接带隙与间接带隙的差值ΔEg < 0.25 eV, 杂化泛函计算的带隙$ {{E}}_{\text{g}}^{\text{HSE}} $ 为1.0—1.8 eV.最后, 针对已筛选出的化合物, 进一步计算了材料的SLME和声子色散谱, 最终筛选出10个SLME > 30%且动力学稳定(声子振动模式无虚频)的化合物.
-
第一性原理计算使用VASP (Vienna abinitio simulation package)软件包[48]进行. 采用广义梯度近似(generalized gradient approximation, GGA)中的PBE(Perdew-Burke-Ernzerhof )[49]泛函来计算交换关联能. 电子-离子相互作用通过投影缀点平面波(projected augmented wave, PAW)[50]赝势进行描述. 所有化合物的平面波基组的能量截断设定为450 eV. K点选择为Γ-center的6×6×6的K点网格. 在几何优化过程中, 晶格单元及其内部原子都被完全弛豫, 直到每个原子上的剩余力小于0.01 eV/Å. 能量收敛标准为10–5 eV. 为了获得准确的能带结构, 采用了包含25%的Hartree-Fock交换关联能的HSE (Heyd-Scuseria-Ernzerhof)[51]杂化泛函. 光学性质的计算使用PBE泛函, 并使用剪刀算符将带隙调整至HSE水平. Ehull的计算使用Pymatgen的Phase Diagram模块[52,53], 并根据Material Project给出的参考文献[54]进行修正. 声子谱的计算通过Phonopy软件包[55,56]辅助VASP进行, 我们将原胞扩展至2×2×2的超晶胞, 并使用有限位移法计算声子色散. 电子有效质量计算使用了Vaspkit[57]软件包进行处理.
-
先对这些新生成的A2M2M'Q4化合物的能量进行计算, 并根据它们的化学式使用材料数据库Materials Project确定可能的分解产物, 并使用其数据计算出Ehull. Ehull描述了具有相同化学成分的二次相之间的竞争关系[58,59], 也就是一种材料在这种化学成分下分解为一组最稳定的材料需要的能量. 例如, 对于四元化合物A2M2M'Q4, 它的Ehull则可以表示为:
$E_{\mathrm{hull}}=E(A_2M_2M'Q_4)-E_{\mathrm{total}} $ (competing phases), 其中E(A2M2M'Q4)为四元化合物A2M2M'Q4的能量,$E_{\mathrm{total}} $ (competing phases)为与A2M2M'Q4化合物具有相同的化学成分的二次相线性组合的最低能量值. 通常情况下, Ehull > 0的化合物有分解为更稳定的化合物的趋势, 而Ehull > 0.05 eV/atom的化合物在实验上通常难以合成[60,61]. 因此, 我们只考虑具有热力学稳定(Ehull = 0)和亚稳定(0 < Ehull < 0.05 eV/atom)的材料.在9种已报道的化合物中, 8种材料稳定的结构与实验一致, 如表1所示, 说明本工作采取的筛选策略是合理的. 对于Na2Cu2ZrS4, 我们计算得到的新稳定相为Ibam, 其能量低于实验的C2/m结构, 这两种结构的Ehull差值为0.06 eV/atom.
接下来, 对材料的带隙值进行计算. 在高通量计算中, 快速地估计材料的带隙对于加速搜索目 标化合物至关重要. 因此, 选择使用GGA-PBE泛函计算这类硫族化合物的带隙(
$ {{E}}_{\text{g}}^{\text{PBE}} $ ), 如图3(a)所示, 计算结果显示这类化合物的带隙范围在0—2.4 eV之间. 由于PBE泛函对带隙的低估[62,63], 因此在这一步的筛选中选择$ {{E}}_{\text{g}}^{\text{PBE}} $ 在0.4—1.2 eV范围内的化合物, 从而筛选出了202种化合物.最小直接带隙
$ {{E}}_{\text{g}}^{\text{direct}} $ 和带隙Eg之间的差异值ΔEg是一个衡量非辐射复合损失的重要指标. 由于直接带隙的特性(ΔEg = 0), 直接带隙半导体在光伏领域备受欢迎. 然而, 一些间接带隙材料(ΔEg > 0)也可以是优秀的光伏材料, 例如Si和MAPbI3, 尽管它们的光吸收和载流子激发效率不如直接带隙材料高, 但这种差距可以通过增大吸收层厚度来弥补[64], 这表明ΔEg < 0.25 eV的材料也可以是良好的太阳能电池吸收层[65]. 我们从PBE计算得到的能带结构中分析了这202种化合物的ΔEg. 如图3(b)所示, ΔEg < 0.25 eV的化合物为148种. 然后, 采用杂化泛函HSE06对这148种材料的电子结构进行了计算, 得到更加准确的带隙值. 从而筛选出72种具有合适的带隙值(1.0 eV <$ {{E}}_{\text{g}}^{\text{HSE}} $ < 1.8 eV)的化合物. 表1中HSE06计算得到的带隙值与实验值符合得很好, 证明了HSE06计算的可靠性. -
接下来, 基于Shockley-Queisser理论[66], 计算这72种化合物在AM1.5 G照明条件下的SLME[67]. 如图4(a)所示, 这72种化合物的SLME与
$ {{E}}_{\text{g}}^{\text{HSE}} $ 之间的关系表明, Ibam和P2/c空间群的化合物更接近Shockley-Queisser极限, 这是因为Ibam和P2/c空间群的材料具有合适的带隙 (1.0—1.6 eV), 带隙差异较小(ΔEg < 0.08 eV). 而其他3种空间群的化合物, 由于有较大的ΔEg和较差的吸收光谱, 因此效率不如前两种空间群. P42/mcm空间群的材料在这5种空间群的材料中效率最低, 主要是因为它们的ΔEg较大(平均为0.19 eV), 导致了较大的非辐射复合损失. 另一方面, C222空间群的化合物的效率比P42/mcm空间群更高, 可能是由于它们相对较小的ΔEg (平均为0.14 eV). 总体而言, Ibam和P2/c类型的材料表现出较高的效率, 共有13种化合物的SLME超过30%.为了进一步评估这13种材料的光伏应用前景, 通过计算声子色散谱来确定其动力学稳定性. 动力学稳定性反映了系统对原子位移的容忍度[68], 可以通过观察声子谱中是否有虚频来判断. 图4(b)和补充材料图S1 (
online )给出了这13种材料的声子色散谱. 从图中可以看出, 其中有7种材料没有虚频, 表明它们具有良好的动力学稳定性. 有3种材料具有显著的虚频, 表明它们是不稳定的. 另外3种材料具有较小的虚频, 表明它们是亚稳定的. 补充材料表S1(online )给出了10种动力学稳定或亚稳定候选材料的带隙和Ehull等信息. 根据晶体结构的统计分析, 这些SLME超过30%的化合物都是Ibam和P2/c空间群的, 表明这两种空间群的材料比本研究中其他空间群的材料具有更好的光伏性能. 从组成的角度来看, 这10中材料中有9种材料是碲化物, 1种硒化物, M' 位上的元素均为IVA族元素而没有IVB族元素. 希望这些统计结果可以为高性能光伏材料的设计提供灵感.图4(c), (d)展示了SLME超过31%的4种材料的SLME与厚度的关系和吸收光谱. 这4种材料分别是Ibam-Rb2Ag2GeTe4 (31.8%), Ibam-Rb2Ag2SnTe4 (31.6%), P2/c-Rb2Cu2SnTe4 (31.1%)和P2/c-Rb2Cu2GeTe4 (31.1%). 相比之下, 这4种候选材料的吸收性能优于CZTS, 在较小的厚度下其SLME就可以达到最高值的90%, 说明这类材料在薄膜太阳能电池中具有良好的应用前景.
-
为了更深入地理解这类材料的电子结构与光学性能, 以评估该类材料的光伏应用价值, 我们进一步对Ibam-Rb2Ag2GeTe4与P2/c-Rb2Cu2GeTe4的HSE06能带结构和载流子有效质量进行了计算. 如图5(a)所示, Ibam-Rb2Ag2GeTe4是一种带隙为1.27 eV的间接带隙半导体, 其导带底(CBM)位于Γ点, 其价带顶(VBM)位于Γ点和X点之间. 但其带隙差异ΔEg只有约0.01 eV, 略低于MAPbI3的ΔEg值(0.03 eV)[69], 因而具有良好的光学性能. 态密度图显示其价带顶附近由Ag-4d和Te-5p轨道贡献, 导带底附近由Ge-4s和Te-5p轨道贡献. 由于Rb的离子性较强, 其在费米能级附近没有明显的贡献. 可以看出, Ibam-Rb2Ag2GeTe4和CBTS具有类似的电子结构, 其主要原因是两种化合物中均保留了[MQ4]和[M'Q4]四面体结构单元. 同时, 与CBTS类似, 碱金属Rb+离子的加入, 预期可以有效改善材料的缺陷性质[29,30]. 同时, 从能带图还可以看出, 导带显示出比价带更大的色散, 意味着电子的输运性质比空穴更好. 这与电子和空穴有效质量的计算结果一致. 从图5(a)可以看出, 该材料具有与二维钙钛矿类似的层状结构, 因此材料的有效质量在层内与层间呈现出较为明显的各向异性. 对于电子来说, 其有效质量较小(0.13m0—0.57m0), 说明电子沿不同方向都表现出良好的输运特性. 而空穴在层内的有效质量(约0.3m0)远小于层间(约14.7m0), 意味着空穴在层内也具有良好的输运特性. 因此, 在利用该材料制备薄膜太阳能电池器件时, 需要考虑光生载流子沿层内方向抽取. 图5(b)给出了P2/c-Rb2Cu2GeTe4的计算结果. 从能带图可以看出该材料是一种带隙为1.18 eV的直接带隙半导体, 它的CBM和VBM都处在Γ点. 同样该材料的电子结构也与CBTS类似. 态密度图显示Rb对CBM和VBM均没有明显贡献. VBM主要由Cu-3d和Te-5p轨道贡献, CBM主要由Ge-4s和Te-5p轨道贡献. 这种材料的各向异性相对于Ibam-Rb2Ag2GeTe4更弱, 有可能是层间的相互作用更强所导致. 其输运性质与Ibam-Rb2Ag2GeTe4类似, 即电子在各个方向均表现出良好的输运性质, 而空穴在层内具有良好的输运性质. 本工作筛选出的其他SLME超过31%具有P2/c和Ibam相的材料, 也呈现出类似的电子结构和载流子输运性质, 如补充材料图S4(
online )所示. 为了进一步了解电子相互作用, 我们还计算了这4种化合物的晶体轨道哈密顿布局函数(COHP)[70], 如补充材料图S5 (online )所示. 费米能级附近的价带顶主要由反键态(—COHP < 0)组成, 导带底附近也表现出明显的反键态. 这种价带和导带边缘附近出现的反键态表明了它们可能具有良好的缺陷容忍度[71,72], 这种特性可以抑制由缺陷引起的非辐射载流子复合, 使得这种太阳能电池吸收层在存在缺陷的情况下仍然保持其良好的性能[73,74]. -
本工作提出了一种基于第一性原理计算和高通量搜索的筛选策略, 以评估四元硫族化合物A2M2M'Q4的电子结构与光学性质. 通过对5种原型结构进行元素替换, 建立了一个包含1350种A2M2M'Q4化合物的材料数据库. 第一性原理计算表明543种材料具有良好的热力学稳定性, 在此基础上, 针对具有光伏应用前景的材料进行筛选, 最终确定了10种太阳能电池吸收层的候选材料. 计算结果表明, 这些候选材料具有良好的热力学稳定性(Ehull < 0.01 eV/atom)、理想的带隙(1.0—1.5 eV)、微小的带隙差异(ΔEg < 0.04 eV) 、较高的可见光吸收系数(104—106 cm–1)以及较低的载流子有效质量(
$m^* < m_0 $ ), 其SLME均超过30%. 它们的电子结构和光学性质与CZTS材料相似, 但由于引入了离子性较强的碱金属阳离子, 可能展现出比CZTS更高的缺陷容忍度. 这些发现为开发更高效、更稳定的太阳能电池材料提供了新的方向. 未来的研究可以进一步探索这些材料的实际制备和器件性能, 以验证其在实际应用中的可行性. 我们计算得到的A2M2M'Q4化合物数据集也能成为将来机器学习等工作的数据基础. -
本篇论文的关联数据可以在科学数据银行
https://www.doi.org/10.57760/sciencedb.j00213.00006 中获取.感谢上海大学高效能计算中心、上海智能计算系统工程技术研究中心(项目编号: 19DZ2252600)提供的计算资源和技术支持.
新型四元硫族化合物光伏特性的高通量筛选和第一性原理研究
Photovoltaic properties of novel quaternary chalcogenides based on high-throughput screening and first-principles calculations
-
摘要: 本工作提出了一种对Cu2ZnSnS4中Zn元素异价取代策略, 探讨了新型四元硫族化合物A2M2M'Q4 (A = Na, K, Rb, Cs, In, Tl; M = Cu, Ag, Au; M' = Ti, Zr, Hf, Ge, Sn; Q = S, Se, Te)作为新型太阳能电池吸收层材料的应用潜力. 利用第一性原理高通量计算, 评估了1350种A2M2M'Q4化合物热力学稳定性、带隙、光谱极限最大效率和声子色散谱等特性. 结果表明, 有10种热力学和动力学稳定的候选材料, 它们表现出合适的带隙, 并展现出高的光吸收性能, 光谱极限最大效率的理论值均超过30%. 它们的电子结构和光学性质类似于Cu2ZnSnS4, 有望应用于高效单结薄膜太阳能电池. 本文数据集可在
https://www.doi.org/10.57760/sciencedb.j00213.00006 中访问获取.Abstract:In recent decades, the demand for clean energy has promoted extensive research on solar cells as a key renewable energy source. Among the various emerging absorber layer materials, Kesterite-type semiconductors have aroused significant interest. Especially, Kesterite Cu2ZnSnS4 (CZTS) stands out as a promising candidate for low-cost thin-film solar cells due to its direct bandgap, high optical absorption coefficient, suitable bandgap (1.39–1.52 eV), and abundance of constituent elements. However, the power conversion efficiency (PCE) of CZTS-based solar cells currently lags behind that of Cu(In,Ga)Se2 (CIGS) cells, mainly due to insufficient open-circuit voltage caused by a large number of disordered cations and defect clusters, resulting in non-radiative recombination and band-tail states. To address these challenges, partial or complete cation substitution has become a viable strategy for altering the harmful defects in CZTS. This study proposes a heterovalent substitution of Zn in CZTS and explores the potential of novel quaternary chalcogenide compound A2M2M'Q4 (A = Na, K, Rb, Cs, In, Tl; M = Cu, Ag, Au; M' = Ti, Zr, Hf, Ge, Sn; Q = S, Se, Te) as absorbers for solar cells. By substituting elements in five prototype structures, a comprehensive material database comprising 1350 A2M2M'Q4 compounds is established. High-throughput screening and first-principles calculations are used to evaluate the thermodynamic stabilities, band gaps, spectroscopic limited maximum efficiencies (SLMEs), and phonon dispersions of these compounds. Our research results indicate that 543 compounds exhibit thermodynamic stability (Ehull < 0.01 eV/atom), 202 compounds possess suitable band gaps (1.0–1.5 eV), and 10 compounds meet all the criteria for thermodynamic and dynamic stability, suitable band gaps, and high optical absorption performance (104–106 cm–1), with theoretical SLME values exceeding 30%. Notably, Ibam-Rb2Ag2GeTe4 exhibits the highest SLME (31.8%) in these candidates, featuring a band gap of 1.27 eV and a small carrier effective mass (< m0). The electronic structures and optical properties of these compounds are comparable to those of CZTS, which makes them suitable for highly efficient single-junction thin-film solar cells. All the data presented in this work can be found at https://www.doi.org/10.57760/sciencedb.j00213.00006 . -

-
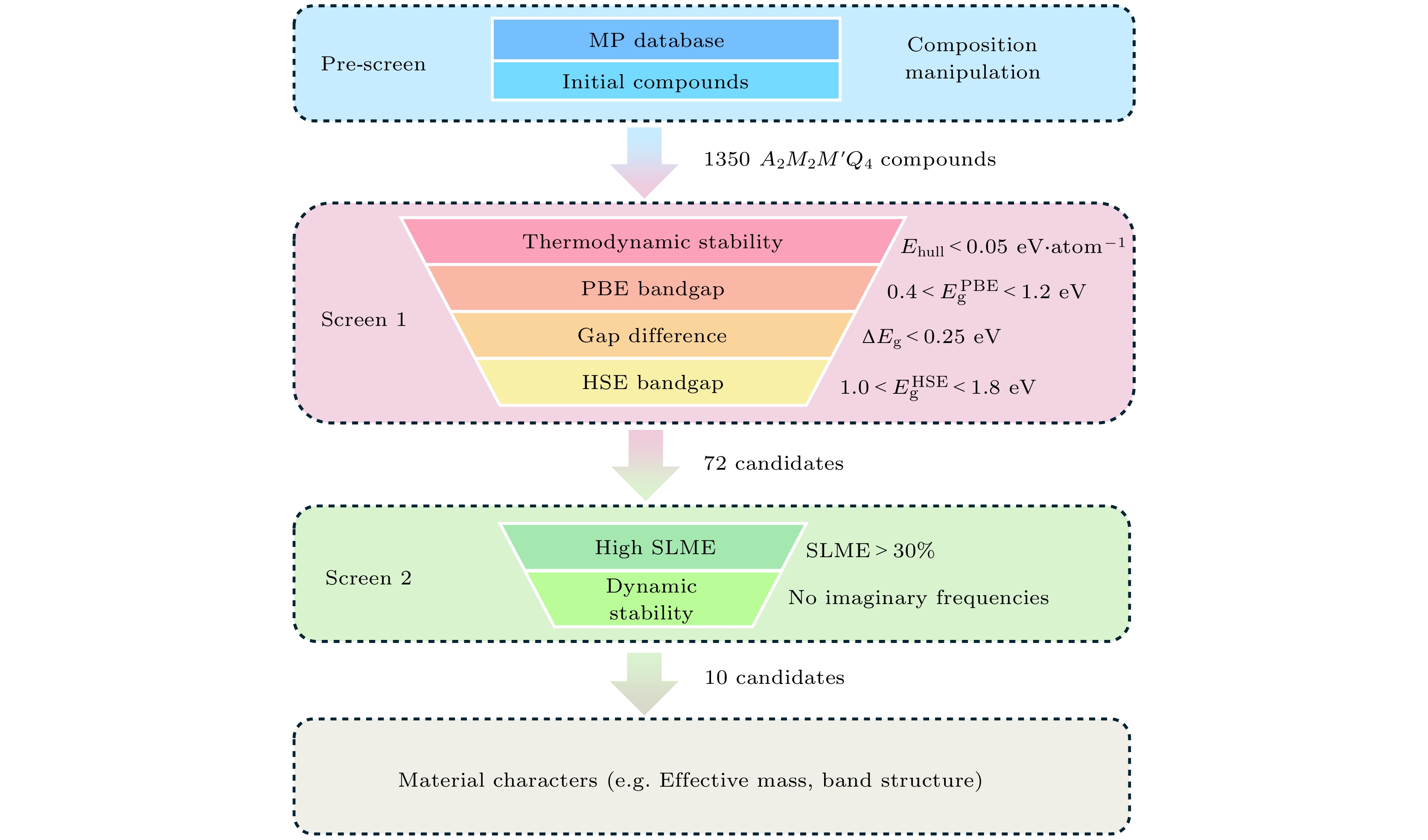
图 2 使用高通量第一性原理计算筛选新材料的工作流程示意图
Figure 2. Schematic workflow of novel materials’ discovery with high-throughput first principle calculations.
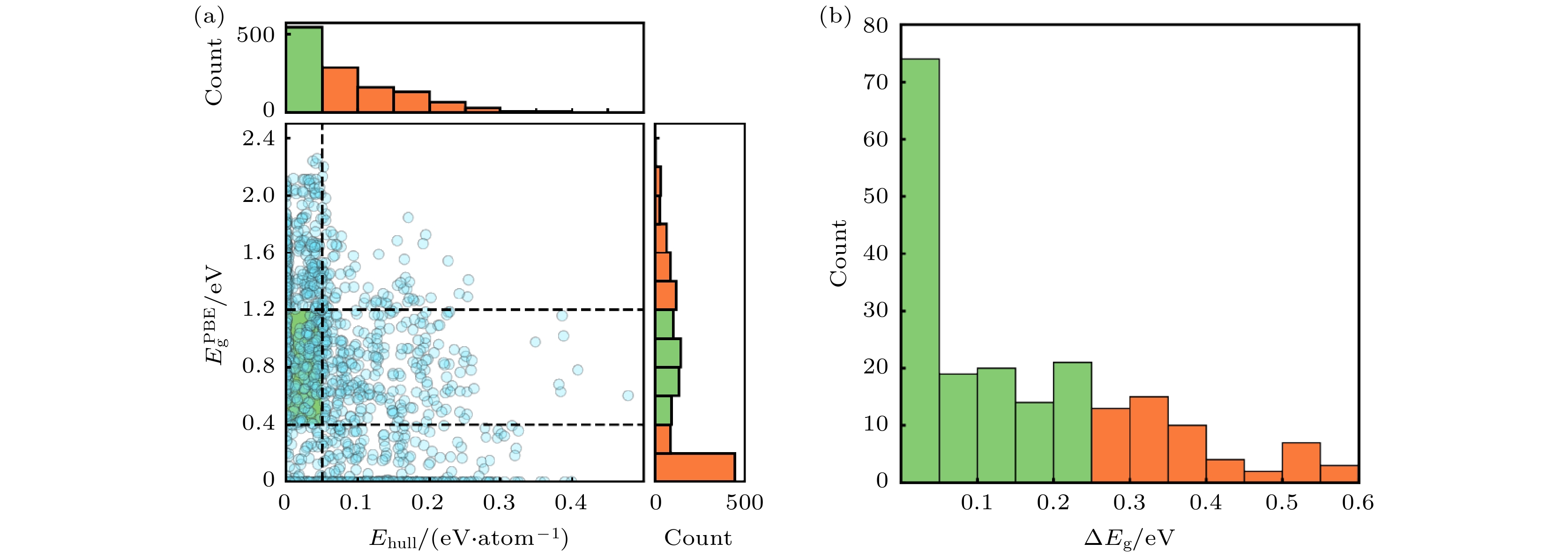
图 3 (a) 1350种A2M2M'Q4化合物的Ehull和
$ {{E}}_{\text{g}}^{\text{PBE}} $ 的散点图和直方图; (b) 202种化合物的ΔEg直方图Figure 3. (a) The scatter plot and histograms of Ehull and
$ {{E}}_{\text{g}}^{\text{PBE}} $ for 1350 A2M2M'Q4 compounds; (b) histogram of ΔEg for 202 compounds.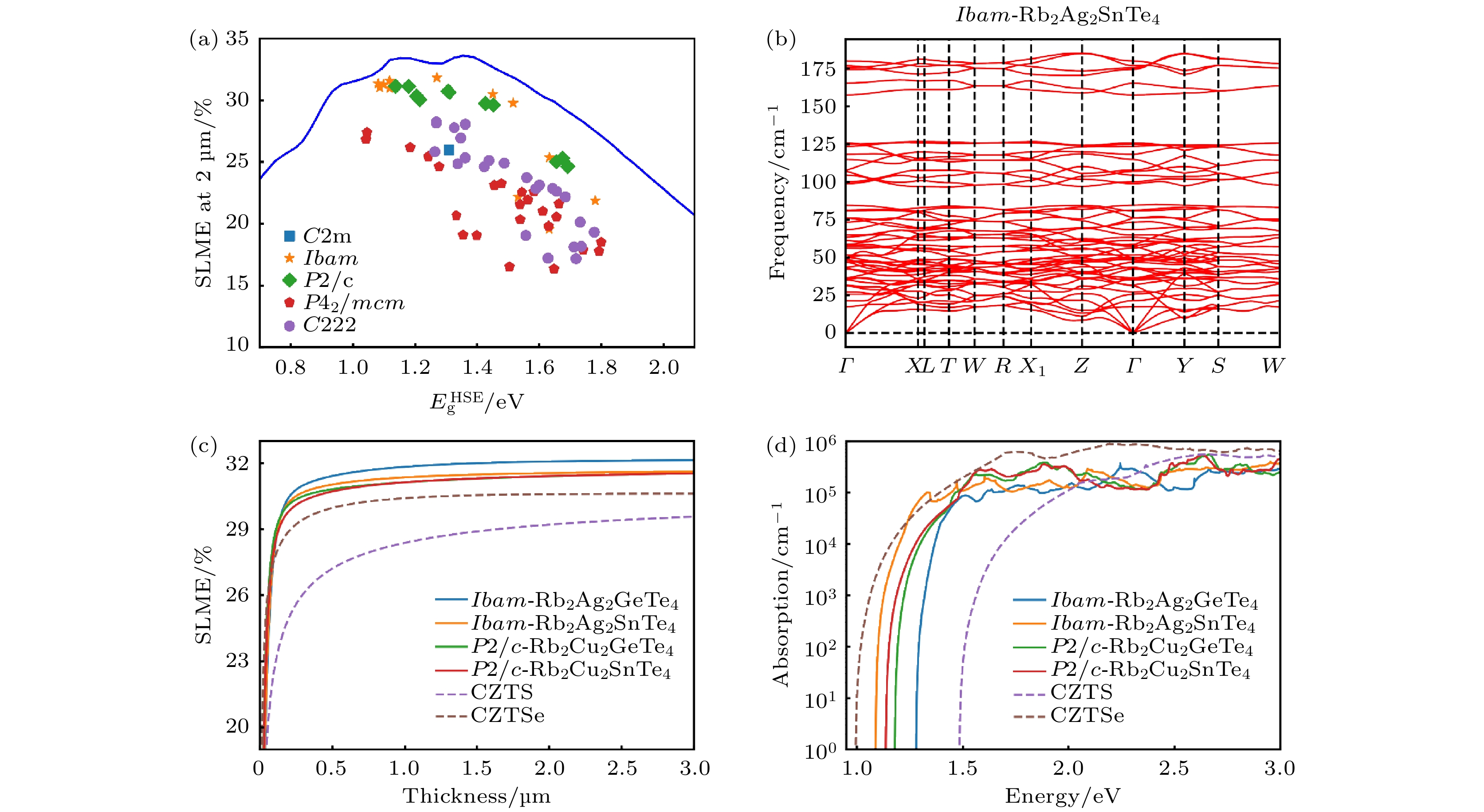
图 4 (a) 72种化合物在薄膜厚度为2 μm时的SLME与
$ {{E}}_{\text{g}}^{\text{HSE}} $ 的关系图, 蓝线为Shockley-Queisser极限; (b) Ibam-Rb2Ag2SnTe4的声子色散谱; 4种SLME超过31%的候选材料, CZTS和CZTSe的(c) SLME与薄膜厚度的关系和(d) 光吸收系数Figure 4. (a) SLME at 2 μm vs. the HSE bandgap
$ {{E}}_{\text{g}}^{\text{HSE}} $ for the 72 compounds, the blue curve represents the Shockley-Queisser limit; (b) phonon dispersion of Ibam-Rb2Ag2SnTe4; (c) thickness dependent SLME values and (d) optical absorption spectra of the top 4 compounds (SLME > 31%), CZTS and CZTSe.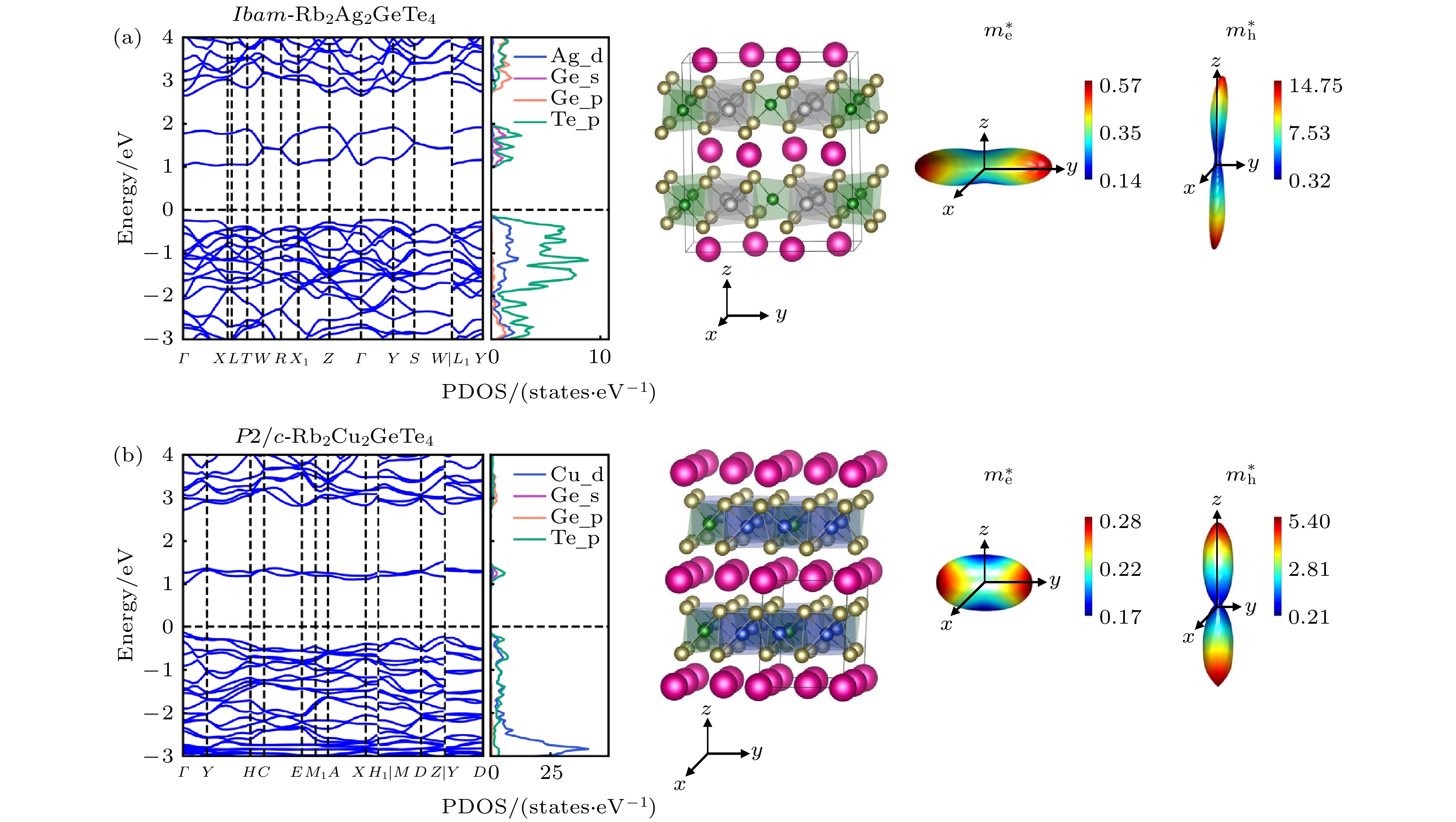
图 5 (a) Ibam-Rb2Ag2GeTe4; (b) P2/c-Rb2Cu2GeTe4的能带结构、分态密度、晶体结构和载流子有效质量
Figure 5. Band structure, partial density of states (PDOS), structure and effective mass of (a) Ibam-Rb2Ag2GeTe4; (b) P2/c-Rb2Cu2GeTe4.
表 1 四元硫族化合物A2M2M'Q4实验报道的带隙和结构与本工作的对比
Table 1. Reported structures and bandgap for A2M2M'Q4 systems.
Compounds Ehull/(eV·atom–1) Stable phase Eg/eV Experiment This work Experiment HSE06 Na2Cu2ZrS4[43] 0.129 C2/m Ibam — 0.07 Cs2Ag2ZrTe4[44] 0 C222 C222 — 2.08 Rb2Cu2SnS4[38] 0.012 Ibam Ibam 2.08 2.02 K2Ag2SnSe4[41] 0 P2/c P2/c 1.8 1.69 Cs2Ag2TiS4[45] 0.001 P42/mcm P42/mcm 2.44 2.44 Cs2Cu2TiS4[45] 0 P42/mcm P42/mcm — 2.56 K2Cu2TiS4[45] 0.002 P42/mcm P42/mcm 2.04 2.62 Rb2Ag2TiS4[45] 0.002 P42/mcm P42/mcm 2.33 2.45 Rb2Cu2TiS4[45] 0.001 P42/mcm P42/mcm 2.19 2.63  下载: 导出CSV
下载: 导出CSV
-
[1] Gloeckler M, Sankin I, Zhao Z 2013 IEEE J. Photovolt. 3 1389 doi: 10.1109/JPHOTOV.2013.2278661 [2] Sobayel K, Shahinuzzaman M, Amin N, Karim M R, Dar M A, Gul R, Alghoul M A, Sopian K, Hasan A K M, Akhtaruzzaman M 2020 Sol. Energy 207 479 doi: 10.1016/j.solener.2020.07.007 [3] Zhou J Z, Xu X, Wu H J, Wang J L, Lou L C, Yin K, Gong Y C, Shi J J, Luo Y H, Li D M, Xin H, Meng Q B 2023 Nat. Energy 8 526 doi: 10.1038/s41560-023-01251-6 [4] Zhang Z F, Yuan X, Lu Y S, He D M, Yan Q H, Cao H Y, Hong F, Jiang Z M, Xu R, Ma Z Q, Song H W, Xu F 2024 Acta Phys. Sin. 73 098803 doi: 10.7498/aps.73.20240153 [5] Wang J, Chen H, Wei S H, Yin W J 2019 Adv. Mater. 31 1806593 doi: 10.1002/adma.201806593 [6] Keller J, Kiselman K, Donzel Gargand O, Martin N M, Babucci M, Lundberg O, Wallin E, Stolt L, Edoff M 2024 Nat. Energy 9 467 doi: 10.1038/s41560-024-01472-3 [7] Todorov T K, Tang J, Bag S, Gunawan O, Gokmen T, Zhu Y, Mitzi D B 2013 Adv. Energy Mater. 3 34 doi: 10.1002/aenm.201200348 [8] Wang K, Gunawan O, Todorov T, Shin B, Chey S J, Bojarczuk N A, Mitzi D, Guha S 2010 Appl. Phys. Lett. 97 143508 doi: 10.1063/1.3499284 [9] Mitzi D B, Gunawan O, Todorov T K, Wang K, Guha S 2011 Sol. Energy Mater. Sol. Cells 95 1421 doi: 10.1016/j.solmat.2010.11.028 [10] Niki S, Contreras M, Repins I, Powalla M, Kushiya K, Ishizuka S, Matsubara K 2010 Prog. Photovoltaics 18 453 doi: 10.1002/pip.969 [11] Chen S Y, Walsh A, Gong X G, Wei S H 2013 Adv. Mater. 25 1522 doi: 10.1002/adma.201203146 [12] Shin D, Saparov B, Mitzi D B 2017 Adv. Energy Mater. 7 1602366 doi: 10.1002/aenm.201602366 [13] Chen S Y, Yang J H, Gong X G, Walsh A, Wei S H 2010 Phys. Rev. B 81 245204 doi: 10.1103/PhysRevB.81.245204 [14] Rey G, Larramona G, Bourdais S, Choné C, Delatouche B, Jacob A, Dennler G, Siebentritt S 2018 Sol. Energy Mater. Sol. Cells 179 142 doi: 10.1016/j.solmat.2017.11.005 [15] Gershon T, Lee Y S, Antunez P, Mankad R, Singh S, Bishop D, Gunawan O, Hopstaken M, Haight R 2016 Adv. Energy Mater. 6 1502468 doi: 10.1002/aenm.201502468 [16] Gong Y C, Qiu R C, Niu C Y, Fu J J, Jedlicka E, Giridharagopal R, Zhu Q, Zhou Y G, Yan W B, Yu S T, Jiang J J, Wu S X, Ginger D S, Huang W, Xin H 2021 Adv. Funct. Mater. 31 2101927 doi: 10.1002/adfm.202101927 [17] Chagarov E, Sardashti K, Kummel A C, Lee Y S, Haight R, Gershon T S 2016 J. Chem. Phys. 144 104704 doi: 10.1063/1.4943270 [18] Yuan Z K, Chen S Y, Xiang H, Gong X G, Walsh A, Park J S, Repins I, Wei S H 2015 Adv. Funct. Mater. 25 6733 doi: 10.1002/adfm.201502272 [19] Zhang J, Liao J, Shao L X, Xue S W, Wang Z G 2018 Chin. Phys. Lett. 35 083101 doi: 10.1088/0256-307X/35/8/083101 [20] Su Z, Tan J M R, Li X, Zeng X, Batabyal S K, Wong L H 2015 Adv. Energy Mater. 5 1500682 doi: 10.1002/aenm.201500682 [21] Bao W, Sachuronggui, Qiu F Y 2016 Chin. Phys. B 25 127102 doi: 10.1088/1674-1056/25/12/127102 [22] Yan C, Sun K, Huang J, Johnston S, Liu F, Veettil B P, Sun K, Pu A, Zhou F, Stride J A, Green M A, Hao X 2017 ACS Energy Lett. 2 930 doi: 10.1021/acsenergylett.7b00129 [23] Luan H M, Yao B, Li Y F, Liu R J, Ding Z H, Zhang Z Z, Zhao H F, Zhang L G 2021 J. Alloy. Compd. 876 160160 doi: 10.1016/j.jallcom.2021.160160 [24] Su Z H, Liang G X, Fan P, Luo J T, Zheng Z H, Xie Z G, Wang W, Chen S, Hu J G, Wei Y D, Yan C, Huang J L, Hao X J, Liu F Y 2020 Adv. Mater. 32 2000121 doi: 10.1002/adma.202000121 [25] Wang C C, Chen S Y, Yang J H, Lang L, Xiang H J, Gong X G, Walsh A, Wei S H 2014 Chem. Mater. 26 3411 doi: 10.1021/cm500598x [26] Shin D, Saparov B, Zhu T, Huhn W P, Blum V, Mitzi D B 2016 Chem. Mater. 28 4771 doi: 10.1021/acs.chemmater.6b01832 [27] Ge J, Yu Y, Yan Y F 2016 ACS Energy Lett. 1 583 doi: 10.1021/acsenergylett.6b00324 [28] Chen Z, Sun K W, Su Z H, Liu F Y, Tang D, Xiao H R, Shi L, Jiang L X, Hao X J, Lai Y Q 2018 ACS Appl. Energ. Mater. 1 3420 doi: 10.1021/acsaem.8b00514 [29] Hong F, Lin W, Meng W W, Yan Y F 2016 Phys. Chem. Chem. Phys. 18 4828 doi: 10.1039/C5CP06977G [30] Xiao Z W, Meng W W, Li J V, Yan Y F 2017 ACS Energy Lett. 2 29 doi: 10.1021/acsenergylett.6b00577 [31] Zhu T, Huhn W P, Wessler G C, Shin D, Saparov B, Mitzi D B, Blum V 2017 Chem. Mater. 29 7868 doi: 10.1021/acs.chemmater.7b02638 [32] Teymur B, Kim Y, Huang J, Sun K, Hao X, Mitzi D B 2022 Adv. Energy Mater. 12 2201602 doi: 10.1002/aenm.202201602 [33] Du Y C, Wang S S, Tian Q W, Zhao Y C, Chang X H, Xiao H Q, Deng Y Q, Chen S Y, Wu S X, Liu S Z 2021 Adv. Funct. Mater. 31 2010325 doi: 10.1002/adfm.202010325 [34] Kuo D H, Tsega M 2014 Jpn. J. Appl. Phys. 53 035801 doi: 10.7567/JJAP.53.035801 [35] Sun Q Z, Shi C, Xie W H, Li Y F, Zhang C X, Wu J H, Zheng Q, Deng H, Cheng S Y 2024 Adv. Sci. 11 2306740 doi: 10.1002/advs.202306740 [36] Maeda T, Kawabata A, Wada T 2015 Phys. Status Solidi. Conf. 12 631 doi: 10.1002/pssc.201400345 [37] Chen S, Gong X G, Walsh A, Wei S H 2009 Phys. Rev. B 79 165211 doi: 10.1103/PhysRevB.79.165211 [38] Liao J H, Kanatzidis M G 1993 Chem. Mater. 5 1561 doi: 10.1021/cm00034a029 [39] Löken S, Tremel W 1998 Z. Anorg. Allg. Chem. 624 1588 doi: 10.1002/(SICI)1521-3749(199810)624:10<1588::AID-ZAAC1588>3.0.CO;2-X [40] Li J, Guo H Y, Proserpio D M, Sironi A 1995 J. Solid State Chem. 117 247 doi: 10.1006/jssc.1995.1270 [41] Chen X A, Huang X Y, Fu A H, Li J, Zhang L D, Guo H Y 2000 Chem. Mater. 12 2385 doi: 10.1021/cm0000447 [42] An Y, Baiyin M, Liu X, Ji M, Jia C, Ning G 2004 Inorg. Chem. Commun. 7 114 doi: 10.1016/j.inoche.2003.10.014 [43] Mansuetto M F, Ibers J A 1995 IEEE J. Solid-State Circuit 117 30 doi: 10.1006/jssc.1995.1242 [44] Pell M A, Ibers J A 2002 J. Am. Chem. Soc. 117 6284 doi: 10.1021/ja00128a016 [45] Huang F Q, Ibers J A 2001 Inorg. Chem. 40 2602 doi: 10.1021/ic001346d [46] Sun B H, He J Q, Zhang X, Bu K J, Zheng C, Huang F Q 2017 J. Alloy. Compd. 725 557 doi: 10.1016/j.jallcom.2017.07.187 [47] Jain A, Ong S P, Hautier G, Chen W, Richards W D, Dacek S, Cholia S, Gunter D, Skinner D, Ceder G, Persson K A 2013 APL Mater. 1 011002 doi: 10.1063/1.4812323 [48] Hafner J 2008 J. Comput. Chem. 29 2044 doi: 10.1002/jcc.21057 [49] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 doi: 10.1103/PhysRevLett.77.3865 [50] Blöchl P E 1994 Phys. Rev. B 50 17953 doi: 10.1103/PhysRevB.50.17953 [51] Paier J, Marsman M, Hummer K, Kresse G, Gerber I C, Ángyán J G 2006 J. Chem. Phys. 124 154709 doi: 10.1063/1.2187006 [52] Ong S P, Wang L, Kang B, Ceder G 2008 Chem. Mater. 20 1798 doi: 10.1021/cm702327g [53] Ong S P, Jain A, Hautier G, Kang B, Ceder G 2010 Electrochem. Commun. 12 427 doi: 10.1016/j.elecom.2010.01.010 [54] Wang A, Kingsbury R, McDermott M, Horton M, Jain A, Ong S P, Dwaraknath S, Persson K A 2021 Sci Rep 11 15496 doi: 10.1038/s41598-021-94550-5 [55] Togo A 2022 J. Phys. Soc. Jpn. 92 012001 doi: 10.7566/JPSJ.92.012001 [56] Togo A, Chaput L, Tadano T, Tanaka I 2023 J. Phys. : Condens. Matter 35 353001 doi: 10.1088/1361-648X/acd831 [57] Wang V, Xu N, Liu J C, Tang G, Geng W T 2021 Comput. Phys. Commun. 267 108033 doi: 10.1016/j.cpc.2021.108033 [58] Jin H, Zhang H, Li J, Wang T, Wan L, Guo H, Wei Y 2019 J. Phys. Chem. Lett. 10 5211 doi: 10.1021/acs.jpclett.9b01977 [59] Singh A K, Montoya J H, Gregoire J M, Persson K A 2019 Nat. Commun. 10 443 doi: 10.1038/s41467-019-08356-1 [60] Liu Y T, Li X B, Zheng H, Chen N K, Wang X P, Zhang X L, Sun H B, Zhang S 2021 Adv. Funct. Mater. 31 2009803 doi: 10.1002/adfm.202009803 [61] Sun W, Dacek S T, Ong S P, Hautier G, Jain A, Richards W D, Gamst A C, Persson K A, Ceder G 2016 Sci. Adv. 2 e1600225 doi: 10.1126/sciadv.1600225 [62] Morales García Á, Valero R, Illas F 2017 J. Phys. Chem. C 121 18862 doi: 10.1021/acs.jpcc.7b07421 [63] Tran F, Blaha P 2009 Phys. Rev. Lett. 102 226401 doi: 10.1103/PhysRevLett.102.226401 [64] Zeng L, Yi Y, Hong C, Liu J, Feng N, Duan X, Kimerling L C, Alamariu B A 2006 Appl. Phys. Lett. 89 111111 doi: 10.1063/1.2349845 [65] Gan Y, Miao N, Lan P, Zhou J Z, Elliott S R, Sun Z 2022 J. Am. Chem. Soc. 144 5878 doi: 10.1021/jacs.1c12620 [66] Shockley W, Queisser H J 1961 J. Appl. Phys. 32 510 doi: 10.1063/1.1736034 [67] Yu L, Zunger A 2012 Phys. Rev. Lett. 108 068701 doi: 10.1103/PhysRevLett.108.068701 [68] Wang V, Tang G, Liu Y C, Wang R T, Mizuseki H, Kawazoe Y, Nara J, Geng W T 2022 J. Phys. Chem. Lett. 13 11581 doi: 10.1021/acs.jpclett.2c02972 [69] Zheng F, Tan L Z, Liu S, Rappe A M 2015 Nano Lett. 15 7794 doi: 10.1021/acs.nanolett.5b01854 [70] Deringer V L, Tchougréeff A L, Dronskowski R 2011 J. Phys. Chem. A 115 5461 doi: 10.1021/jp202489s [71] Yang D W, Lv J, Zhao X G, Xu Q L, Fu Y H, Zhan Y Q, Zunger A, Zhang L J 2017 Chem. Mater. 29 524 doi: 10.1021/acs.chemmater.6b03221 [72] Yamada Y, Nakamura T, Endo M, Wakamiya A, Kanemitsu Y 2015 IEEE J. Photovolt. 5 401 doi: 10.1109/JPHOTOV.2014.2364115 [73] Pandey M, Rasmussen F A, Kuhar K, Olsen T, Jacobsen K W, Thygesen K S 2016 Nano Lett. 16 2234 doi: 10.1021/acs.nanolett.5b04513 [74] Wexler R B, Gautam G S, Stechel E B, Carter E A 2021 J. Am. Chem. Soc. 143 13212 doi: 10.1021/jacs.1c05570 -
计量
- 文章访问数: 763
- HTML全文浏览数: 763
- PDF下载数: 9
- 施引文献: 0