
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
活性物质可以通过消耗内部能量或吸收周围环境中的能量来实现自我驱动. 这种自我驱动和能量的持续输入导致活性物质系统远离平衡, 从而表现出一系列新奇的动力学行为, 例如运动诱导相分离或大规模集体运动[1,2]等. 人们通过理论、模拟和实验的方法, 致力于探索各种活性物质的非平衡现象.
“活性聚合物”是重要的一大类活性物质. 其尺度不一, 广泛存在于各种生命系统中. 例如真核细胞中的染色质[3,4]、细胞骨架中的微管和微丝[5,6]、细长细菌[7,8]、藻类[9]、蠕虫[10]等. 除了生物活性聚合物, 在实验上还可以通过多种方式将人工合成的自推进胶体组合成活性聚合物, 例如利用高离子浓度下的强引力使Janus粒子形成链状结构, 通过调节外加电场的频率控制自推进的速度和方向[11]. 当系统中有大量活性聚合物时, 空间相互作用与聚合物的构象自由度相结合会引起很多集体行为和新奇的自组装现象. 形状、相互作用、推进方式、环境和边界条件等许多因素都在集体行为中起着重要作用. 例如在二维空间中, 中等密度下的切向驱动细丝在不同持续长度和推进力大小的影响下出现了熔融态、气态分布的螺旋和移动的团簇态等, 在更高密度时形成堵塞、向列排列和活性湍流[12]. 在微管和分子马达混合物的体外实验中, 马达动态交联微管并沿微管定向运动, 导致微管形成涡旋结构和星形结构[13]. 超过临界密度时, 细丝自组织成团簇、涡流和互相连接的条带[14,15]等. 此外, 向微管和驱动蛋白混合物中添加耗尽剂会导致微管间产生有效吸引从而组装成数百微米长的束(bundles)[16,17]. 在足够高的浓度下, 微管束会形成逾渗活性凝胶(percolating active network). 这种凝胶网络具有流体动力不稳定性、增强运输和流体混合等与细胞生物学高度相关的动力学特性[16,18,19]. 自然界中很多可以看作活性聚合物的生物体也可以通过界面张力等有效吸引作用自组装形成活性聚合物凝胶, 比如蓝藻[20,21]和秀丽隐杆线虫[22].
在非活性的聚合物体系中, 由ABA三嵌段共聚物形成的物理缔合凝胶(其中A嵌段间具有缔合作用)也受到了广泛的关注. 在嵌段共聚物溶液中, 分子内结合和分子间结合相互竞争, 两种结合的比例随温度和浓度而变化. 因此, 除了不与任何链结合的自由链, ABA共聚物有3种可能的构象[23]: 1)环(loop), 即末端两个A嵌段属于同一微畴; 2)桥(bridge), 即末端嵌段连接两个不同的微畴; 3)悬空(dangling), 即一个末端嵌段不属于任意微畴. 当ABA 三嵌段共聚物溶解到中间嵌段选择性溶剂中时, 优先形成环状结构并结合成胶束[24]. 随着共聚物浓度的增大, 一部分中间嵌段会桥接不同胶束的核心, 最终导致逾渗网络的形成, 网络与溶剂共同构成凝胶. 目前对活性体系中嵌段共聚物的研究刚刚起步, 只关注链上的驱动力分布不均匀带来的影响[25,26], 单体之间相互作用是一致的. 我们曾报道过A嵌段之间具有吸引作用的自驱动AB二嵌段共聚物自组装[27], 发现不同的驱动力和吸引强度可以使链表现出丰富的结构和动力学行为. 本文建立了活性ABA三嵌段共聚物模型, 并探究其自组装行为.
本文我们感兴趣的问题是自主驱动的存在如何影响ABA 三嵌段共聚物的凝胶化转变以及凝胶的结构与动力学. 研究结果表明活性ABA共聚物的凝胶态有3种类型: 稳定聚合物凝胶、动态聚合物凝胶和塌缩聚合物凝胶. 逾渗通路和链构象的稳定性与A嵌段之间的吸引强度有关, 凝胶的空间均匀性主要受活性驱动的影响. 在凝胶网络的拓扑结构方面, 当A嵌段之间吸引较强时, 环链比例随活性力增强而增加, 当吸引强度较小时情况则相反. 而交联点的分支数随活性的变化除了受到吸引强度的影响, 还与链的刚性有关, 通常半柔性链网络的分支更多. 在动力学方面, 活性聚合物的定向运动会引发稳定聚合物凝胶整体的反常扩散. 本文的研究不仅有助于增进对活性聚合物集体行为的认识, 也有望为高分子材料在生物医药、材料工程等领域的设计和应用提供参考.
-
采用分子动力学模拟来研究活性ABA共聚物的集体自组装. 嵌段共聚物的两端A嵌段各由
${N_{\text{A}}}$ 个A单体组成, 中间B嵌段由${N_{\text{B}}}$ 个B单体组成, 聚合度为${N_{\text{l}}} = \left( {2{N_{\text{A}}} + {N_{\text{B}}}} \right)$ . 所有单体模拟成质量为$m$ 、直径为$\sigma $ 的粒子, 粒子之间的非键相互作用由LJ势表示:其中
$r$ 表示两个非键单体之间的距离,$\alpha $ 和$\beta $ 表示两个相互作用的单体的类型,${\varepsilon _{\alpha \beta }}$ 和$r_{\alpha \beta }^{\text{c}}$ 分别表示单体之间的相互作用强度和相互作用的截断半径.${\varepsilon _{\alpha \beta }}$ 和$r_{\alpha \beta }^{\text{c}}$ 的选择决定了A单体和B单体之间的异质性, 如图1所示. 为了模拟溶液中的两亲共聚物链, 假设A嵌段是疏水的, 倾向于塌缩聚集, 而B嵌段是亲水的, 倾向于在溶液中膨胀. 因此将A-B和B-B单体之间的有效非键相互作用设置为纯排斥的WCA(Weeks-Chandler-Anderson)势, 即相互作用势在$r_{{\text{AB}}}^{\text{c}} = r_{{\text{BB}}}^{\text{c}} = {2^{1/6}}\sigma $ 处截断, 并设置相互作用强度${\varepsilon _{{\text{AB}}}} = {\varepsilon _{{\text{BB}}}} = 1$ . 而非键的A单体之间存在有效吸引, 即$r_{{\text{AA}}}^{\text{c}} = 2.5\sigma $ , 其相互作用强度$ {\varepsilon _{{\text{AA}}}} = \varepsilon $ 是可调节的, 用于量化非键的A单体之间的吸引强度. 此外不再考虑单体与溶剂分子之间的相互作用.在链内, 相邻单体之间通过弹簧势连接:
其中
${{\boldsymbol{r}}_i}$ 是聚合物链上第$i$ 个单体的位置向量,$ {{\boldsymbol{r}}_{i, i + 1}} = {{\boldsymbol{r}}_{i + 1}} - {{\boldsymbol{r}}_i} $ 表示相邻两个单体之间的键向量.${r_0}$ 表示键的平衡长度,${k_{\text{s}}}$ 表示弹簧势系数. 相邻键之间的键角弯曲势为其中
$\kappa $ 为反应链的刚性弯曲势系数,${\theta _i}$ 是单体$i - 1$ ,$i$ 和$i + 1$ 之间的夹角度数.每个单体的运动由郎之万方程来描述:
等式右边第1项表示的是单体受到的保守力, 其中
${U_i}$ 表示系统中第$i$ 个单体受到的总势能, 包括非键相互作用势、键伸缩势和键角弯曲势. 第2项是单体受到的阻尼力,$\gamma $ 表示平动摩擦系数. 最后一项表示热噪声,${{\boldsymbol{\eta}} _i}\left( t \right)$ 为单位方差高斯白噪声. 每条聚合物链的头部和尾部单体不受活性力的驱动, 而非头尾单体受到的活性力为其中, 单位向量
${\hat {\boldsymbol{t}}_i} = \dfrac{{{{\boldsymbol{r}}_{i - 1}} - {{\boldsymbol{r}}_{i + 1}}}}{{\left| {{{\boldsymbol{r}}_{i - 1}} - {{\boldsymbol{r}}_{i + 1}}} \right|}}$ 表示自驱动力沿活性链的切线方向[27,28].采用 LAMMPS 软件进行模拟, 模拟中设置
$m = 1$ ,$\sigma = 1$ 和${k_{\text{B}}}T = 1$ 作为质量、长度和能量约化单位, 其中${k_{\text{B}}}$ 是玻尔兹曼常数,$T$ 为温度. 因此时间约化单位为$\tau = \sqrt {m{\sigma ^2}/({k_{\text{B}}}T)} $ . 每条链由${N_{\text{l}}} = 30$ 个单体构成, 其中${N_{\text{A}}} = 5$ ,${N_{\text{B}}} = 20$ . 系统中活性链的数目为$N = 500$ , 二维盒子的边长$L = 200$ , 面密度$\rho = N{N_{\text{l}}}{\sigma ^2}{\text{π }}/{(2 L)^2} = 0.3$ , 模拟采用周期性边界条件. 设置键的平衡长度${r_0} = 1$ , 弹簧势系数${k_{\text{s}}} = 2000{k_{\text{B}}}T/{\sigma ^2}$ 以减少链长波动的影响. 另外设置摩擦系数$\gamma = 10$ 以确保活性链的运动是在过阻尼环境下进行的[27]. 在正式模拟之前, 将活性力关闭, 并将所有单体之间的非键相互作用设置为WCA势, 进行充分的热平衡. 随后打开活性力并改变单体之间的对相互作用进行模拟, 时间步长为$0.001\tau $ , 总模拟时长为$5 \times {10^4}\tau $ . -
图2给出了纯柔性系统(
$\kappa = 0$ )中通过调整活性力大小和A嵌段吸引强度所形成的6种相, 根据是否发生逾渗转变划分出两种类型: 凝胶态和非凝胶态(补充材料(online) ). 凝胶态中包括稳定聚合物凝胶(stable polymer gel, SPG)、动态聚合物凝胶(dynamic polymer gel, DPG)、塌缩聚合物凝胶(collapsed polymer gel, CPG), 非凝胶态包括熔融态(melt)、团簇态(clusters)和螺旋态(spirals).吸引作用的存在使得分子间和分子内的A嵌段会发生聚集. 如果两个A嵌段内有单体质心之间的距离小于吸引作用范围, 则认为这两个A嵌段属于同一个吸引团簇(红色微畴). 吸引团簇可以充当网络的交联点, 通过桥链与多个吸引团簇相连, 最终聚集成全空间无限延伸的逾渗网络, 也就是聚合物凝胶. 如果逾渗几率超过0.5, 就可认为发生了凝胶化转变.
当吸引较大、活性力较小时, 系统会发生凝胶化转变. 此时吸引团簇内的A嵌段紧密结合, 链的构象和逾渗通路比较稳定, 把这种状态称为稳定聚合物凝胶(图3(a)). 随着活性力增大, 吸引的影响逐渐减小. 原本被限制在网络中的链开始增强运动, 导致网络塌缩成一个内部结构紧密并且逾渗的大团簇, 把这种逾渗网络称为塌缩聚合物凝胶(图3(b)和补充材料Movie 1
(online) ). 当活性力更大时, 原本逾渗的塌缩大团簇也会分解成小团簇, 整个系统转变为不逾渗的团簇态(图3(c)).随着吸引强度逐渐减弱, A嵌段之间的结合变得松散、不稳定. 当吸引强度减小到
$\varepsilon = 2$ 时, 小活性系统中仍然能发生凝胶化转变. 但弱吸引和小活性驱动使得网络链的构象和位置不断发生变化, 因此逾渗通路也会发生变化, 这种状态被称为动态聚合物凝胶(图3(d), 补充材料Movie 2(online) ). 当吸引强度减小到$\varepsilon = 1$ 时, 柔性系统已经不会发生凝胶化转变, 活性力较小时嵌段共聚物大多呈现与非活性均聚物类似的熔融态(图3(e)). 活性力较大时, 大多数链围绕自身头部缠绕起来并旋转, 呈现出强自驱动引发的螺旋结构, 这种状态称为螺旋态(图3(f)).图4展示了3种凝胶态的不同特点. 凝胶网络中的链可以有3种构象: 自身两端A嵌段结合的环链、两端分别与不同链的A嵌段结合的桥链以及有一端不与任何A嵌段结合的悬空链, 此外还有一种不与任何链连接的自由链. 图4(a)统计了系统中每条链在
$1 \times {10^4}\tau $ 以内维持一个构象的平均时间$\left\langle {{T_{{\text{con}}}}} \right\rangle = \displaystyle \sum\nolimits_T {({T^2}{n_T})} \Big/\sum\nolimits_T {(T{n_T})} $ , 其中$T$ 表示某条链连续保持一个构象的时间,${n_T}$ 表示维持时间为$T$ 的次数. 图4(a)中, 动态聚合物凝胶($\varepsilon = 2$ )链的构象维持时间总是较短的, 吸引强度增大后构象维持时间与统计时间几乎一致. 图4(b)是3种典型凝胶态中, 参与逾渗的链数与总链数比值随时间的变化. 对于吸引较强、交联点结合稳固的稳定凝胶态, 几乎系统中所有链都参与构成凝胶网络, 且比例不随时间变化. 在吸引较弱、链的构象经常变化的动态凝胶态中, 构成网络的链数随时间总是具有一定的波动, 甚至偶尔会有一小段时间不逾渗. 在塌缩凝胶态中, 构成逾渗网络的链数相对动态凝胶更稳定, 但也会出现一小段时间不逾渗的情况. -
在A嵌段之间施加较强的吸引可以促使不同链迅速结合, 这种结合使得演化初期就会出现逾渗网络. 活性力较小时, 一旦链通过A嵌段间的结合形成网络, 那么自驱动就会受到网络整体的牵制, 局部的定向运动不明显. 当活性力较大但不足以形成螺旋时, 系统中大部分链同样也会很快形成网络, 只是网络中的链间结合对自驱动能力的限制变小了. 因此吸引团簇内A嵌段的无规堆积所产生的力矩可以带动周围的链进行旋转, 网络局部位置的运动变强, 引发了塌缩(补充材料 Movie 1
(online) ).图5展示了
${F_{\text{a}}} = 0.3$ ,$\varepsilon = 7$ 时网络塌缩的过程(补充材料Movie 1(online) ). 活性力引发力矩较大的交联点和周围的链旋转, 并通过桥链带动其他交联点向自身靠拢. 这导致网络中产生张力, 从而使交联薄弱的桥链被拉伸最终断裂, 悬空端就近连接到附近交联点上, 最终形成致密的大团簇. 团簇尺寸较大时可以利用周期性边界条件首尾相接, 保持逾渗状态. -
聚合物网络结构是影响凝胶的机械和光学性能的重要因素, 例如黏弹性、渗透性、拉伸强度、质量传递和光学清晰度等. 通常网络结构并非均匀和规则的, 其不均匀性(inhomogeneities)以跨越多个长度尺度的各种形式表现出来[29]: 在10—100 nm范围, 聚合物网络结构的特点是交联点和链的空间分布不均匀; 在1—10 nm范围, 悬空链、环链和桥链以及三维上的缠结主导了网络结构的大分子特征; 在更小的尺度上, 交联点的分支数是决定网络结构的关键特征.
我们使用Torquato等[30,31]提出的一种简单方法来表示整个网络的空间分布, 以分析其空间均匀性. 首先在网络孔隙区域随机选取一个点, 然后该点为圆心生成一个半径不断扩大的圆, 直到该 圆与任意单体接触, 此时圆的半径
${r_{\text{p}}}$ 反映了该点附近孔隙的大小(补充材料(online) ). 相同聚合物浓度下, 网络对空间的覆盖越均匀, 凝胶的孔隙越小[32,33]. 图6给出了$\varepsilon = 3$ 和$\varepsilon = 7$ 时孔径分布$P({r_{\text{p}}})$ 随活性力大小的变化. 由于随机点不可避免地经常出现在贴近凝胶边缘的位置, 因此峰值总是出现在$0.5\sigma $ 处(截断距离$1\sigma $ –单体半径$0.5\sigma $ ). 在非活性系统中, 空间分布不均匀来源于网络形成过程中浓度波动[29,34]. 在活性系统中, 孔径分布的宽度通常随活性力增加而变宽, 较大的活性导致原本空间分布均匀的网络逐渐塌缩在一起, 交联点和网络链更紧密地分布在团簇内部, 造成空间中出现极大的空洞. 图6(a)中,${F_{\text{a}}} = 0.3$ 的孔径分布比${F_{\text{a}}} = 0.2$ 时偏窄, 图6(b)中,${F_{\text{a}}} = 0.4$ 的孔径分布比${F_{\text{a}}} = 0.3$ 时偏窄, 主要是因为活性增加到塌缩聚合物凝胶和团簇态的相边界时, 容易使凝胶中出现更多非逾渗小团簇或者逾渗团簇的局部位置与整体不贴近, 因此最大孔径变小.网络的拓扑不均匀性指环链、悬空链和局部的多重交联等网络缺陷, 这些缺陷对凝胶的机械性能有很大影响. 图7展示了弱吸引和强吸引情况下, 网络链的3种构象比例随活性变化, 构象比例表示为
$ {P_{{\text{con}}}} = {N_{{\text{con}}}}/{N_{{\text{per}}}} $ , 其中${N_{{\text{con}}}}$ 为某种构象的链数,${N_{{\text{per}}}}$ 为参与逾渗网络的总链数. 图7(a)显示, 当吸引较强时, 环链比例随活性增大而增大, 桥链比例逐渐下降. 这是因为切向驱动柔性链的平均回转半径和平均末端距会随活性增加而下降 [35,28], 这种结构随活性变化的趋势更有利于链内结合. 此外, 由于柔性桥链具有较强的可形变性, 因此在网络塌缩过程中交联点密度较高、网络链更密集的地方会出现桥链两端靠近、两个交联点合并的情况, 此时桥链变环链. 图7(b)为吸引较弱时3种构象的比例, 与吸引较强的情况不同, 从稳定聚合物网络变为塌缩聚合物网络时环链减少而桥链和悬空链增多. 弱吸引时活性共聚物最初也会出现较多环链, 但由于链间结合不稳定, 因此容易受活性驱动、噪声和其他链碰撞的影响而转变构象. 弱吸引的塌缩凝胶态中存在较多的移动团簇, 彼此之间有较频繁的碰撞, 造成环链转变为桥链或悬空链. 图7(c)显示了在$\varepsilon = 3$ 和$\varepsilon = 7$ 的塌缩聚合物凝胶中, 环链比例随时间的变化. 在吸引较弱时, 塌缩聚合物网络的环链比例先增大然后明显下降并在稳态值附近波动.网络中某个交联点可以通过桥链与其他
$f$ 个交联点连接,$f$ 就是该交联点的分支数. 图8展示了不同吸引强度下交联点的分支数分布$P(f) = {N_f}/{N_{{\text{cp}}}}$ 以及交联点尺寸, 即构成交联点的链数分布$P(n) = {N_n}/{N_{{\text{cp}}}}$ , 其中${N_f}$ 是分支数为$f$ 的交联点数目,${N_n}$ 是尺寸为$n$ 的交联点数目,${N_{{\text{cp}}}}$ 是交联点总数目. 不论吸引强弱和活性大小, 交联点链数分布的宽度总是比分支数更宽, 且峰值出现的位置往往比分支数更大, 表明常常存在两个交联点之间有多条桥链相接的情况. 当吸引较弱时($\varepsilon = 3$ ), 活性增大则$f$ 的峰值左移(图8(a)), 塌缩聚合物网络的分支数分布的峰值出现在$f = 1$ , 主要是由于团簇在活性驱动下运动而脱离网络时, 较弱的吸引作用导致交联断裂的链保持悬空. 当吸引较强时($\varepsilon = 7$ ), 很少有交联断裂引发悬空链的情况. 即使有悬空链也会很快连接到其他交联点上, 因此峰值的位置没有明显变化, 基本出现在$f = 3$ 或$f = 4$ 的位置(图8(c)). 强吸引下活性力增大导致分支数峰值减小, 而较大分支数的比例略有增长, 主要是由于活性力引起的网络收缩导致较大尺寸的交联点数目增加. 图8(d)中交联点尺寸分布的变化和分支数分布的变化基本一致.选取弯曲势系数
$\kappa = 5$ 和$\kappa = 100$ 来模拟活性半柔性链和刚性链的凝胶化转变, 以此探究刚性对活性嵌段共聚物凝胶化的影响(补充材料(online) ). 由于一定的刚性有抵抗弯曲的效果, 因此半柔性和刚性凝胶网络中的环链比例大幅下降, 桥链构象占主导. 在半柔性和刚性系统中, 常出现多链平行并列且两端结合的结构, 也就是束. 较大的活性会驱动束沿各自轮廓方向在空间中定向移动, 同样会引发网络的塌缩, 甚至逾渗状态被破坏. 通过计算不同刚性和活性条件下网络的平均交联点尺寸和平均分支数, 我们发现活性和刚性越大则交联点尺寸越大, 但在平均分支数方面则是半柔性网络交联点的分支最多. -
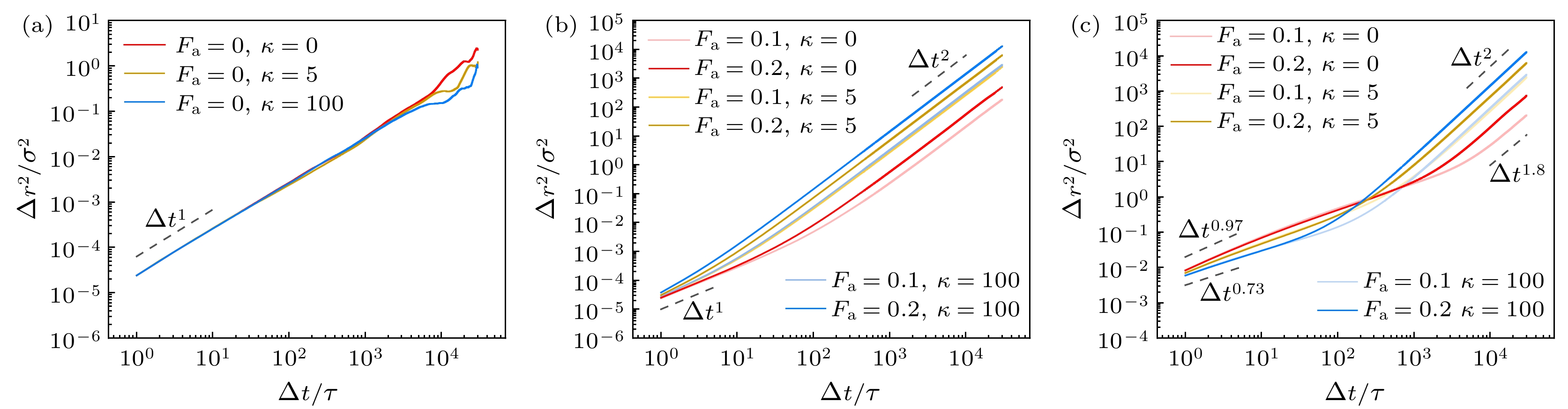
凝胶网络的连接导致活性聚合物的自主驱动力分布在整个网络上, 我们发现稳定聚合物凝胶会发生整体的漂移现象, 这种异常的输运已经在一些理论和实验中有所研究[36-38]. 图9(a), (b)分别为非活性和活性的稳定凝胶整体质心的MSD. 非活性凝胶的整体质心为普通扩散(
$\Delta {r^2}\left( {\Delta t} \right) \sim \Delta {t^\alpha }$ ,$\alpha = 1$ ), 活性凝胶整体质心在长时间尺度下表现为弹道扩散($\alpha = 2$ ), 相同刚性条件下, 活性力越大网络扩散越快. 当活性力相同时, 半柔性和刚性网络的漂移比纯柔性网络更快, 且半柔性和刚性网络MSD相差的数量级很小.图9(c)为活性凝胶网络中每条链质心的MSD. 在短时间尺度内, 网络整体漂移对链质心扩散的影响不显著, 热噪声是链质心扩散的主要因素. 当链的两端通过吸引作用被固定在网络中时, 热噪声的作用效果受到限制, 因此链的质心表现为亚扩散. 从图9(c)可以发现, 短时间内扩散速度只与刚性有关, 与活性大小无关. 纯柔性网络链的扩散速度比半柔性和刚性更快, 是因为柔性系统有很多环链并且桥链不是完全绷直. 在受到吸引作用限制的前提下, 这些链的质心有更宽松的移动空间. 对于半柔性和刚性链来说, 抵抗热噪声引发形变的能力更强, 因此网络中每条链质心的扩散更弱. 在长时间尺度下, 活性力引起的网络整体漂移占主导作用, 因此网络中每条链质心的MSD转变为弹道扩散. 活性力和刚性越大, 链质心扩散的速度越快.
-
关于嵌段共聚物的凝胶化转变往往集中在非活性的平衡体系中, 本文将远离平衡的自驱动性质与ABA 嵌段共聚物结合, 研究了活性ABA嵌段共聚物凝胶在结构和动力学方面的特点. 在二维环境中, 不同的活性和吸引强度下ABA共聚物形成的凝胶态可以分为3种类型: 逾渗通路稳定、空间分布均匀的稳定聚合物凝胶, 逾渗通路和网络链构象不断变化的动态聚合物凝胶, 以及聚集成较大团簇的塌缩聚合物凝胶. 活性凝胶的空间均匀性除了与网络形成过程中的浓度波动有关, 还与活性引起的网络链不一致的运动有关, 表现在柔性系统中交联点的旋转和半柔性及刚性系统中的束沿各自轮廓方向的定向移动. 在网络的拓扑均匀性上, 吸引较低时活性力增加导致环链比例降低, 而吸引较高时, 环链比例随活性增加而提高. 此外增大刚性可以防止环链的形成, 并能够通过排斥体积相互作用诱导定向排序, 导致很多链平行排列成束, 因此交联点的平均尺寸随刚性和活性增大, 但半柔性链形成的网络交联点平均分支数更高. 在动力学上, 活性稳定聚合物凝胶有整体在空间中匀速定向漂移的特征, 且刚性和活性会提升网络漂移的速度.
最后简单讨论了活性嵌段共聚物密度和AB嵌段比例对凝胶化效果的影响(补充材料
(online) ). 增大密度和A嵌段的比例可以使网络抵抗塌缩的能力更强, 逾渗状态不易受活性破坏, 但网络中环链缺陷的比例会增加. 在后续研究中还会继续探究不同参数对活性嵌段共聚物凝胶弹性模量的影响等. 总之, 本文的研究不仅有助于增进对活性聚合物集体行为的认识, 也有望为高分子活性材料的设计和应用提供参考.
活性三嵌段共聚物的凝胶化转变
Gel transition of active triblock copolymers
-
摘要: 活性物质的自推进特征引发了许多非平衡自组织现象, 而聚合物链的构象自由度可以使链产生独特的平衡自组装行为, 这激发了活性物质与聚合物物理的交叉研究. 本文通过分子动力学模拟, 研究了自驱动活性对ABA 型三嵌段共聚物凝胶化转变的调控. 研究结果表明活性嵌段共聚物凝胶的塌缩源于自驱动活性引起的网络链运动, 活性越大则凝胶网络越容易产生大直径空洞. 在凝胶网络的拓扑缺陷方面, 当A嵌段之间吸引强度较大时, 环链比例随活性力增强而增大, 吸引强度较小时情况则相反. 交联点的分支数随活性的变化除了受到吸引强度的影响, 还与链刚性有关. 在动力学方面, 活性聚合物的定向运动会引发稳定聚合物凝胶整体的反常扩散. 本文的研究有助于增进对活性聚合物集体行为的认识, 为高分子活性材料的设计和应用提供了新的思路.Abstract: The self-propulsion of active matter leads to many non-equilibrium self-organization phenomena, and the conformational freedom of polymer chains can produce unique equilibrium self-assembly behaviors, which stimulates cross-disciplinary research between active matter and polymer physics. In this work, we use molecular dynamics simulations to investigate the modulation of self-propulsion activity on the gel transition of ABA triblock copolymers. The research results indicate that under different active forces and attractive strengths, the gel states formed by ABA copolymers can be divided into three types: stable polymer gels with stable percolation paths and uniform spatial distribution, dynamic polymer gels with constantly changing percolation path and strand conformation, and collapsed polymer gels aggregating into large percolating clusters. The spatial uniformity of active gels is related not only to the concentration fluctuation during the formation of the network, but also to the inconsistent movement of the network chains caused by the activity, which is manifested in the rotation of crosslinking points in the flexible system and the directional movement of the bundles along their contour directions in the semi-flexible and rigid systems. In terms of topological conformation of polymer networks, when the attractive strength between A blocks is strong, the proportion of loop increases with the active force increasing. When attractive strength is weak, inter- and intra-chain binding are unstable, and the conformation is easily changed by the activity drive, noise and other chain collisions, so the proportion of loop decreases with the active force increasing. The branching number of crosslinking points varies with active force, which is not only affected by the attraction strength, but also related to the rigidity of the network chain. Generally, the branch number of crosslinking points in semi-flexible networks is larger than that in flexible and rigid networks. In addition, the directional motion of active polymers induces anomalous diffusion in stable polymer gels. This study contributes to the understanding of the collective behavior of active polymers and serves as a guide for designing and implementing active polymeric materials.
-
Key words:
- active matter /
- ABA triblock copolymer /
- non-equilibrium system /
- polymer gel .
-

-
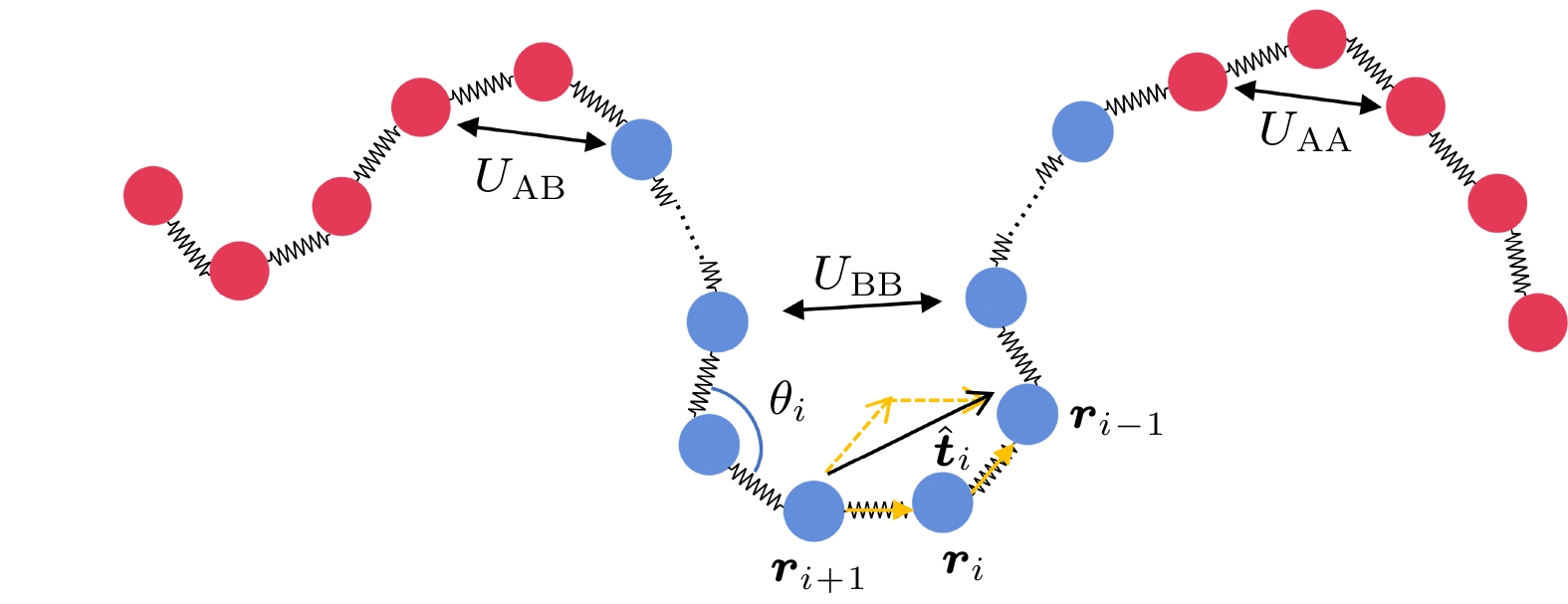
图 1 单体非键相互作用和活性力示意图, 红色圆形表示A单体, 蓝色圆形表示B单体, 中间嵌段的一些B单体在图中被省略
Figure 1. Schematic diagram of non-bonding interactions and active forces of particles, with red circles representing type A monomers and blue circles representing type B monomers, some B monomers in the middle block are omitted in the diagram.
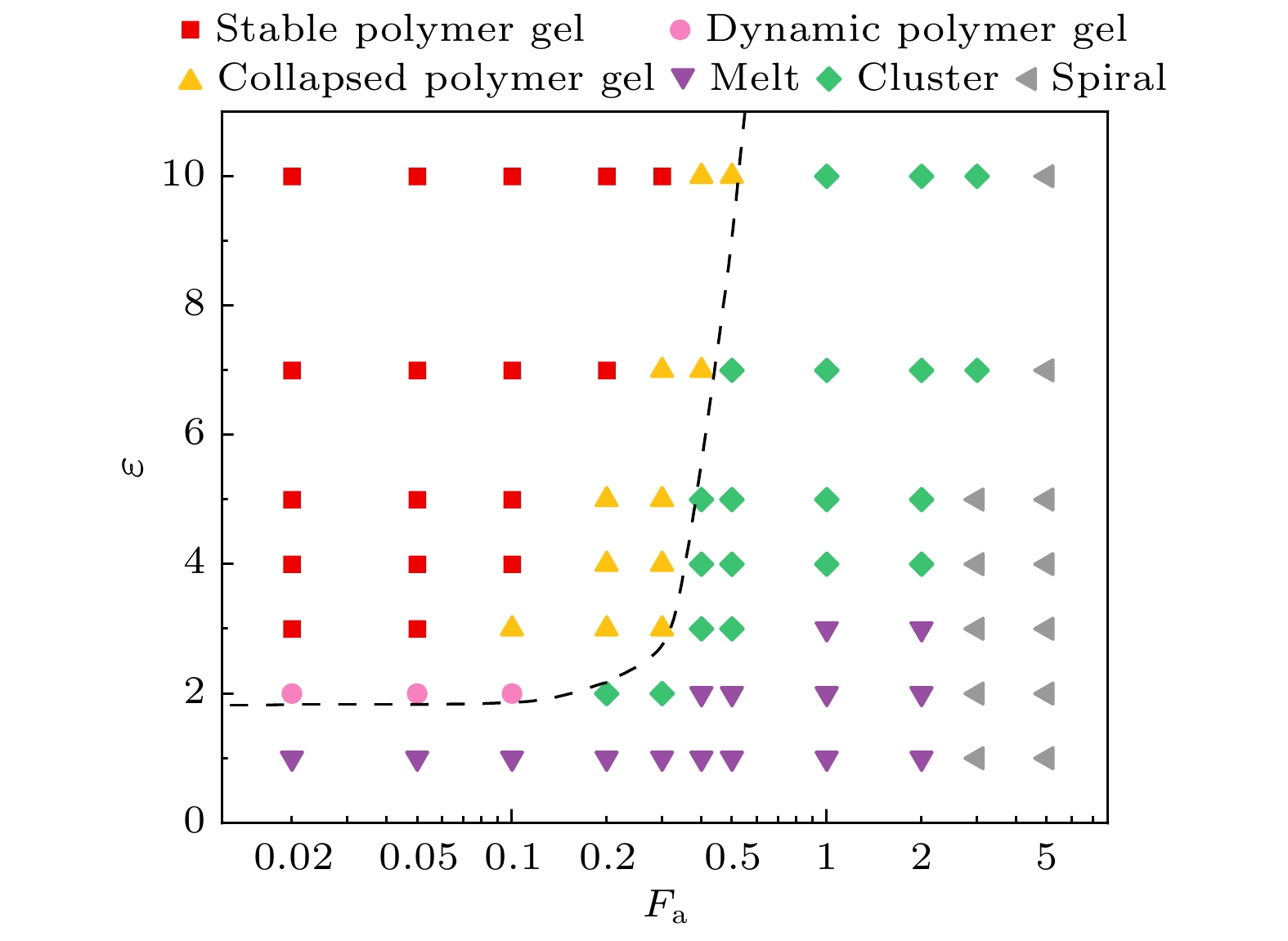
图 2 柔性活性ABA嵌段共聚物的相图, 虚线是各种凝胶态和非凝胶态粗略的分界线, 左侧表示凝胶态, 右侧为非凝胶态
Figure 2. Phase diagram of flexible active ABA block copolymer. The dotted line is the rough dividing line between the various gel states and non-gel states, with the left part representing the gel state and the right part representing the non-gel state.

图 3 柔性活性ABA嵌段共聚物典型相的模拟快照 (a) 稳态聚合物凝胶; (b) 塌缩聚合物凝胶; (c) 团簇态; (d) 动态聚合物凝胶; (e) 熔融态; (f) 螺旋态
Figure 3. Simulated snapshot of typical phase of flexible active ABA block copolymer: (a) Stable polymer gel; (b) collapsed polymer gel; (c) cluster state; (d) dynamic polymer gel; (e) melt state; (f) spiral state.
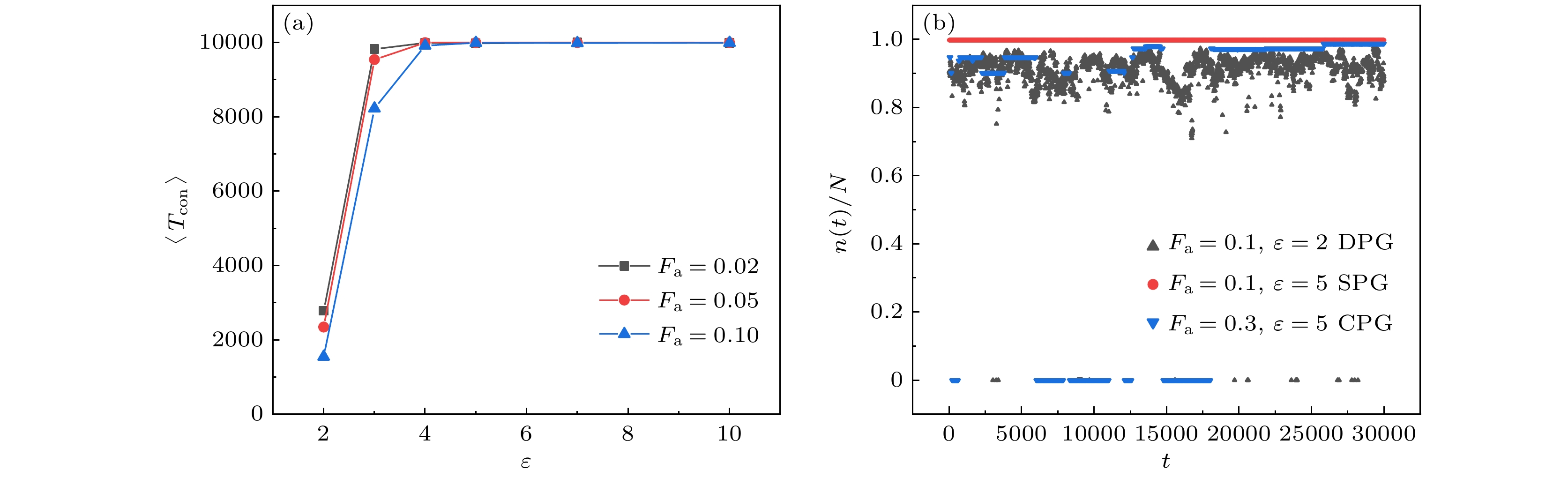
图 4 (a) 参与逾渗的链的构象维持平均时间; (b) 参与逾渗的链数与总链数的比值随时间的变化
Figure 4. (a) The average conformation maintenance time of the chain involved in percolation; (b) the ratio of the number of chains involved in percolation to the total number of chains varies over time.
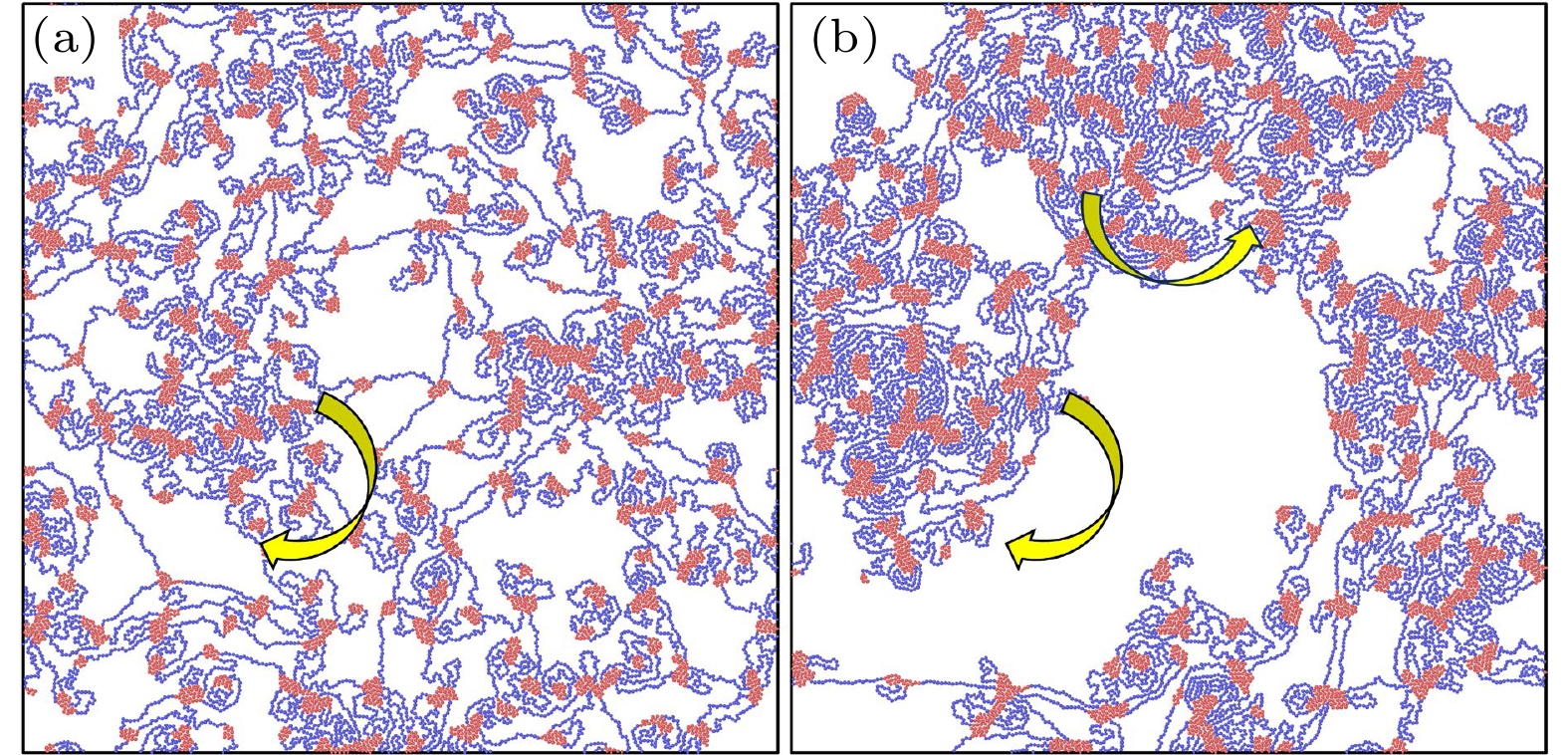
图 5 网络塌缩的演化过程, 黄色箭头表示局部位置的旋转方向 (a)
$t = 200\tau $ ; (b)$t = 2000\tau $ Figure 5. The evolution process of network collapse, with yellow arrows indicating the direction of rotation of the local network: (a)
$t = 200\tau $ ; (b)$t = 2000\tau $ .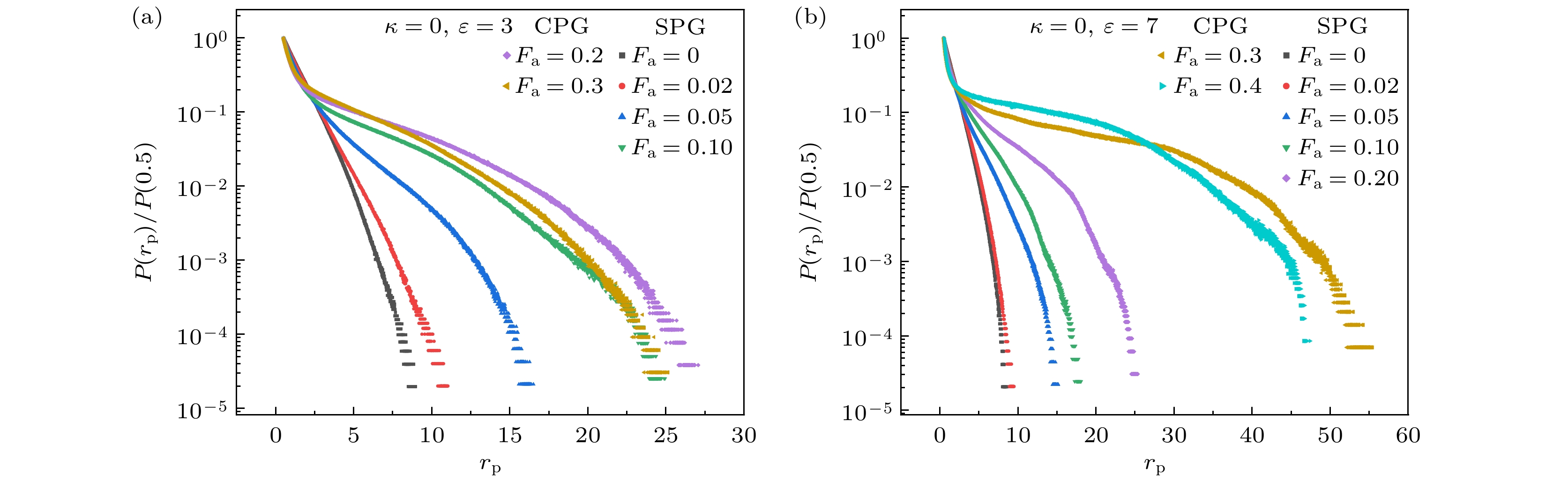
图 6 弱吸引和强吸引条件下, 凝胶的孔径分布 (a)
$\varepsilon = 3$ ; (b)$\varepsilon = 7$ Figure 6. The pore size distribution of the gel with weak and strong attraction strength: (a)
$\varepsilon = 3$ ; (b)$\varepsilon = 7$ .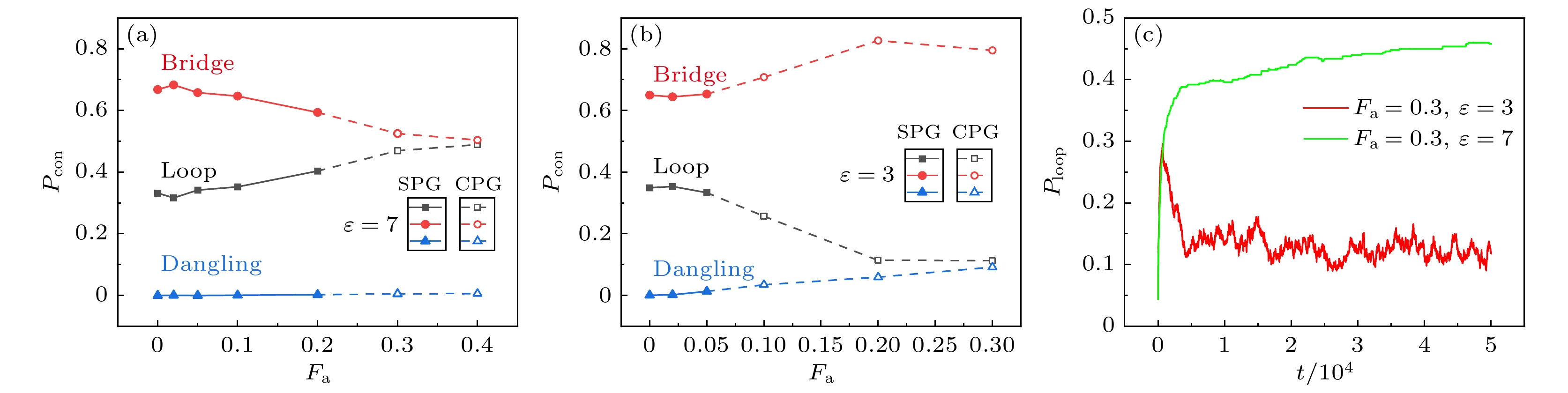
图 7 不同吸引强度下网络链三种构象的比例 (a)
$\varepsilon = 7$ ; (b)$\varepsilon = 3$ ; (c)两种吸引强度的塌缩聚合物凝胶中, 环链比例随时间的变化Figure 7. The proportions of the three conformations of the network chain with different attraction strength: (a)
$\varepsilon = 7$ ; (b)$\varepsilon = 3$ ; (c) the proportion of loop chains in two collapsed polymer gels with different attractive strengths as a function of time.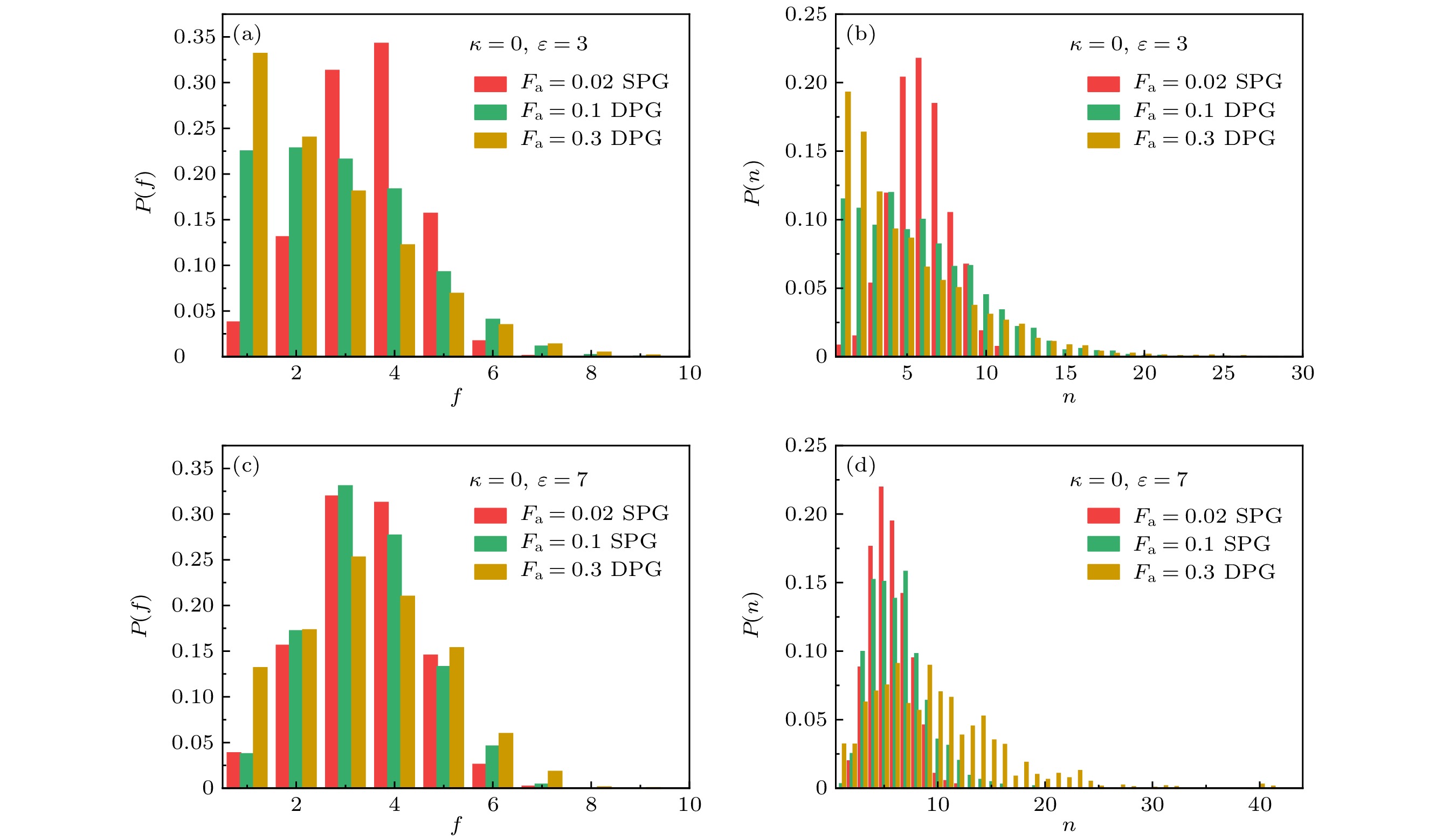
图 8 当
$\varepsilon = 3$ 时, 3种活性下 (a)交联点的分支数分布和(b)交联点的尺寸分布, 红色表示稳定聚合物凝胶, 黄色和绿色表示塌缩; 当$\varepsilon = 7$ 时, 3种活性下(c)交联点的分支数分布和(d)交联点的尺寸分布Figure 8. When
$\varepsilon = 3$ , (a) the distribution of the branch number and (b) the size distribution of the crosslinker under the three activities; when$\varepsilon = 7$ , (c) the distribution of the branch number and (d) the size distribution of the crosslinker under the three activities -
[1] Deseigne J, Dauchot O, Chaté H 2010 Phys. Rev. Lett. 105 098001 doi: 10.1103/PhysRevLett.105.098001 [2] Vicsek T, Zafeiris A 2012 Phys. Rep. 517 71 doi: 10.1016/j.physrep.2012.03.004 [3] Liu L, Shi G, Thirumalai D, Hyeon C 2018 PLoS Comput. Biol. 14 e1006617 doi: 10.1371/journal.pcbi.1006617 [4] Liu K, Patteson A E, Banigan E J, Schwarz J M 2021 Phys. Rev. Lett. 126 158101 doi: 10.1103/PhysRevLett.126.158101 [5] Weber C A, Suzuki R, Schaller V, Aranson I S, Bausch A R, Frey E 2015 Proc. Natl. Acad. Sci. U. S. A. 112 10703 doi: 10.1073/pnas.1421322112 [6] Kučera O, Gaillard J, Guérin C, Théry M, Blanchoin L 2022 Proc. Natl. Acad. Sci. U.S.A. 119 e2209522119 doi: 10.1073/pnas.2209522119 [7] Li H, Shi X Q, Huang M, Chen X, Xiao M, Liu C, Chaté H, Zhang H P 2019 Proc. Natl. Acad. Sci. U. S. A. 116 777 doi: 10.1073/pnas.1812570116 [8] Auer G K, Oliver P M, Rajendram M, Lin T Y, Yao Q, Jensen G J, Weibel D B 2019 mBio 10 00210 doi: 10.1128/mBio.00210-19 [9] Selander E, Jakobsen H H, Lombard F, Kiørboe T 2011 Proc. Natl. Acad. Sci. U. S. A. 108 4030 doi: 10.1073/pnas.1011870108 [10] Deblais A, Prathyusha K R, Sinaasappel R, Tuazon H, Tiwari I, Patil V P, Bhamla M S 2023 Soft Matter 19 7057 doi: 10.1039/D3SM00542A [11] Nishiguchi D, Iwasawa J, Jiang H R, Sano M 2018 New J. Phys. 20 015002 doi: 10.1088/1367-2630/aa9b48 [12] Duman Ö, Isele-Holder R E, Elgeti J, Gompper G 2018 Soft Matter 14 4483 doi: 10.1039/C8SM00282G [13] Ndlec F J, Surrey T, Maggs A C, Leibler S 1997 Nature 389 305 doi: 10.1038/38532 [14] Schaller V, Weber C, Semmrich C, Frey E, Bausch A R 2010 Nature 467 73 doi: 10.1038/nature09312 [15] Huber L, Suzuki R, Krüger T, Frey E, Bausch A R 2018 Science 361 255 doi: 10.1126/science.aao5434 [16] Sanchez T, Chen D T N, DeCamp S J, Heymann M, Dogic Z 2012 Nature 491 431 doi: 10.1038/nature11591 [17] Doostmohammadi A, Ignés-Mullol J, Yeomans J M, Sagués F 2018 Nat. Commun. 9 3246 doi: 10.1038/s41467-018-05666-8 [18] Backouche F, Haviv L, Groswasser D, Bernheim-Groswasser A 2006 Phys. Biol. 3 264 doi: 10.1088/1478-3975/3/4/004 [19] Chew W X, Henkin G, Nédélec F, Surrey T 2023 iScience 26 106063 doi: 10.1016/j.isci.2023.106063 [20] Faluweki M K, Goehring L 2022 J. R. Soc. Interface 19 20220268 doi: 10.1098/rsif.2022.0268 [21] Faluweki M K, Cammann J, Mazza M G, Goehring L 2023 Phys. Rev. Lett. 131 158303 doi: 10.1103/PhysRevLett.131.158303 [22] Sugi T, Ito H, Nishimura M, Nagai K H 2019 Nat. Commun. 10 683 doi: 10.1038/s41467-019-08537-y [23] Li Y Q, Sun Z Y, Shi T F, An L J 2004 J. Chem. Phys. 121 1133 doi: 10.1063/1.1758938 [24] Tanaka F, Koga T 2001 BCSJ 74 201 doi: 10.1246/bcsj.74.201 [25] Hu H X, Shen Y F, Wang C, Luo M B 2022 Soft Matter 18 8820 doi: 10.1039/D2SM01139E [26] Han H, Joo S, Sakaue T, Jeon J H 2023 J. Chem. Phys. 159 024901 doi: 10.1063/5.0150224 [27] Jiao Y, Wang J, Tian W D, Chen K 2023 Soft Matter 19 5468 doi: 10.1039/D3SM00596H [28] Bianco V, Locatelli E, Malgaretti P 2018 Phys. Rev. Lett. 121 217802 doi: 10.1103/PhysRevLett.121.217802 [29] Gu Y, Zhao J, Johnson J A 2020 Angew. Chem. Int. Ed. 59 5022 doi: 10.1002/anie.201902900 [30] Torquato S, Avellaneda M 1991 J. Chem. Phys. 95 6477 doi: 10.1063/1.461519 [31] Agrawal S, Galmarini S, Kröger M 2023 Phys. Rev. E 107 015307 doi: 10.1103/PhysRevE.107.015307 [32] Hosono N, Masubuchi Y, Furukawa H, Watanabe T 2007 J. Chem. Phys. 127 164905 doi: 10.1063/1.2790007 [33] Pereyra R G, Al-Maadeed M A, Carignano M A 2017 Express Polym. Lett. 11 199 doi: 10.3144/expresspolymlett.2017.21 [34] Di Lorenzo F, Seiffert S 2015 Polym. Chem. 6 5515 doi: 10.1039/C4PY01677G [35] Anand S K, Singh S P 2018 Phys. Rev. E 98 042501 doi: 10.1103/PhysRevE.98.042501 [36] Head D A, Gompper G, Briels W J 2011 Soft Matter 7 3116 doi: 10.1039/C0SM00888E [37] Córdoba A 2018 J. Phys. Chem. B 122 4267 doi: 10.1021/acs.jpcb.8b00238 [38] Köhler S, Schaller V, Bausch A R 2011 Nat. Mater 10 462 doi: 10.1038/nmat3009 -
计量
- 文章访问数: 556
- HTML全文浏览数: 556
- PDF下载数: 7
- 施引文献: 0