
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
软物质[1]是泛指在微弱力学作用下和受温度影响下易发生形变的软凝聚态物质, 通常包含有颗粒物质、高分子聚合物、生物物质如生物大分子等. 根据热力学第二定律, 开放的软物质系统会在熵驱动、非共价相互作用以及外力等驱动下发生自组织过程, 形成一定的有序结构. 这种有序结构易受环境因素的影响, 在平衡态和非平衡态情况下均可能存在, 结构稳定性与环境因素通常具有高度关联性. 同时, 在各种调控因素作用和驱动下, 形成的有序微观结构能够在宏观上表现出不同的功能和物理特性. 因此深入探索软物质的自组织物理原理和调控机制与因素, 是研发具有不同功能的软物质新材料的基础. 克服软物质自组织结构和功能的不稳定性及其功能的差异性, 并开发新的软物质自组织构建模块和组装方式是研发软物质新材料的首要挑战.
软物质体系的自组织结构丰富多样, 比如颗粒物质在外场作用下能够形成有序排列[2]、胶体粒子形成有序晶格结构[3,4]、蛋白质分子由于疏水效应等作用形成三维空间的折叠结构[5]、细胞内各种生物分子在熵驱动下形成相分离[6]等. 由生物分子, 包括蛋白质、核酸、多糖等, 构成的基本组件通常呈现出弹性体或凝胶态特征, 进而能够组装成为纳米颗粒结构或水凝胶等材料. 这些相关的生物分子软物质材料由于具有良好的生物相容性, 在生物材料领域被广泛关注, 一直是生物软物质材料研究的前沿课题, 还被应用于肿瘤治疗[7,8]、组织修复[9,10]、细胞支架[11,12]等生物医学领域. 它们通常是由生物分子自组织或与其他单元共组装形成超分子自组装结构. 自然界中, 蛋白质分子构成的超分子自组装结构(或蛋白质复合体结构)尤为常见, 蛋白质分子在弱相互作用驱动下形成热力学稳定的组装结构单元, 并能被环境因素调控而实现多层次组装成为复杂结构或复合结构以行使生命体中多种多样的功能[13,14]. 比如细胞内细胞骨架蛋白的可逆自组装牵引细胞的朝向运动[15], 胶原蛋白自组装成细胞外基质作为细胞支撑骨架[16], 谷胱甘肽S转移酶[17]等大多数水溶性低聚酶与底物特异性地结合. “自下而上”地拆解研究天然蛋白质组装体, 并模仿再现其功能结构, 以期达到更高层次的创新应用是生物分子组装研究领域备受人们关注的缘由. 然而, 天然蛋白质分子组装体一般是由多条链组成(每条链通常由几十到几百甚至千多个氨基酸残基组成, 包含几种或多达20种类氨基酸), 对蛋白质组装体的结构解析和功能动力学过程的了解是十分困难的挑战. 因此, 开发由几个或十几个到几十个氨基酸残基组成的短链多肽分子链及其衍生物作为自组装基元, 并探索由这些基元通过自组装形成的组装结构体及其功能特性——即生物分子软物质功能材料的研究, 已经成为当前的前沿热点课题.
当前的理论研究认为多肽主要通过非共价相互作用, 包括静电相互作用力、氢键、疏水效应、范德瓦耳斯力和π共轭效应等分子间弱相互作用力以实现其自组装. 在整个组装过程中, 通常地, 多肽基元先形成α螺旋或β片等二级结构, 再聚集拼装成纳米管、纳米纤维、纳米球或凝胶等高级复杂结构. 最初形成的二级结构将显著影响其高级结构的物理性质, 比如含有稳定β片的自组装材料具有高机械强度[18]. 同一类型多肽分子构建的简单模块也能发挥多种多样的功能[19], 这是因为多肽自组装的分子间弱相互作用力大多受到氨基酸侧链基团变化、pH、温度、光照等环境因素影响. 由此, 可通过微调氨基酸侧链基团和实验环境参数来直接操控多肽自组装的过程和结构, 以形成不同的高级组装结构并表现出不同的宏观物理性质, 进而完成精细而又复杂的功能材料的构建和设计[14]. 除此以外, 多肽分子可以与其他功能小分子, 如配位金属离子、核酸、酶等同样地依靠非共价相互作用共组装成超分子结构基元[20]. 不同于单一多肽的自组装路径, 超分子组装基元更加灵活, 可以通过工程化多肽和合理搭配各类小分子系统生成更多特殊结构. 最后, 在多肽分子和超分子组装构建块组成的超大材料设计工具箱基础上, 可以引入化学稳定的高分子聚合物得到多层复合凝胶、双网络凝胶等结构复杂、功能特异的新型生物软物质材料, 从而用于靶向药物释放[21]、生物基半导体开发[22]、仿生器官和医用植入传感器制备[23,24]等.
目前, 随着对自组装多肽基元的深入研究以及充分利用多肽分子良好的生物相容性优势, 人们已经研发出具有特殊组织结构、能量耗散牺牲单元或可逆动力学组件的力学性能突出的仿生水凝胶[25]和具有可穿戴功能的柔性电子生物传感器[26,27], 还可结合两者的优势进一步开发出智能仿生软体机器人[28]等器件, 这些方面是该领域的前沿应用发展趋势. 鉴于多肽组装材料日新月异的发展态势, 本文将以多肽分子及其衍生物为典型生物分子, 对自组装生物分子软物质材料领域的发展状况进行了梳理和总结. 从生物分子纳米材料、生物分子凝胶材料和生物分子复合材料三个方面介绍了自组装生物分子软物质材料的分子作用机制、设计原则、构建方法、物理特性以及可能的应用等课题的研究内容和进展, 其中还涉及人工光催化生物平台[29]、压电生物传感器[30]、药物递送水凝胶[31]、人造皮肤[32]等方向. 最后还探讨了基于多肽等生物分子的自组装软物质材料的挑战性课题的研究, 并对该领域的未来发展方向进行探讨和展望.
-
自然界中存在着多种生物分子自组织纳米结构, 比如细胞中各种分子机器的复合结构[33], 阿尔兹海默疾病标志物淀粉样蛋白纤维结构[34]等. 通过探索研究这些天然自组织生物纳米结构和其形成过程以及揭示其生物功能作用等相关特性, 人们受到启发创造出一系列人工合成的生物分子软物质纳米材料. 例如, 通过人工构建一系列不同氨基酸排列序列并合成成为相关分子. 早在20世纪末, Zhang等[35]发现了一种疏水和亲水的氨基酸残基交替排列的短肽, 它能够在疏水作用等弱相互作用下自组装成非常稳定的自互补结构, 进而在宏观上形成具有耐酸、耐碱等特性的薄膜. 在此基础上, 他们还深入研究了离子自互补短肽的特殊结构带来的宏观稳定性[36]、构象转变特性[37], 并探讨了调整氨基酸残基序列的亲疏水性和正负电荷对短肽自组装结构的影响[38], 进而开发出了一系列以自互补短肽为基元的生物分子软物质纳米材料, 可用于哺乳动物细胞的体外黏附机制研究[39]、在组织内创造三维微环境促进细胞再生[40]等. 2011年, 本课题组[41]首次提出了一种可自组装成有序纳米结构的α螺旋型离子自互补短肽. 不同于常见的二级结构为β片的离子自互补肽, 我们精细地调节了短肽的周期性序列和离子对位置, 使得离子自互补短肽以新颖的α螺旋肩对肩组装方式构成扭曲带状原纤微. 同样开辟一类短肽自组装研究的还有Reches和Gazit[42], 他们于2003年首次报道了使用来源于β-淀粉样蛋白的二苯丙氨酸二肽基序(diphenylalanine, FF)能够自组装成稳定的刚性纳米管, 推动了基于FF及其衍生物为代表的超短肽自组织生物分子纳米材料这一课题的蓬勃发展. 该工作引进的方法是将FF从高溶解度有机相分散至低溶解度水相, 仅通过FF之间的非共价相互作用力, 就能够自组装形成肽纳米管. 同时, 改变pH、温度等能够显著影响非共价相互作用力等环境条件, 并调控FF的自组装形态. 以FF为代表, 不改变生物分子的原始序列, 仅通过外界条件介导非共价相互作用力促使生物分子自组装, 进而形成多种纳米形态结构, 如纳米管、纳米球、纳米纤维等, 这是构建自组织生物分子软物质纳米材料的基本方法.
随后, 人们总结了一些对材料的物理化学性质有重要影响作用的特殊化学基团, 进一步筛选出具有特殊功能和性质的常见小分子作为基础构建单元, 尝试将这些构建单元相互拼接以增强材料的特殊物理性质, 如压电特性、高机械强度力学特性等. 含有苯环的天然氨基酸, 如酪氨酸、色氨酸和苯丙氨酸, 因为其苯环具有潜在的π共轭效应和光响应特征, 尤其适合作为高刚度或具有压电响应特征材料的基础构建单元. 如图1(a)所示, Ji等[43]使用溶解度比苯丙氨酸低得多的酪氨酸作为二聚体, 制备了具有紧密堆积成特性的氨基酸晶体. 由于酪氨酸二聚体以截然不同的苯环“面对面”组装方式, 晶体结构内部极化且堆积非常紧密, 使其结构具有优良的机械稳定性和热稳定性以及与传统无机压电材料氧化锌(ZnO)等接近的压电特性. 该工作探索了低成本且易于制备的简单氨基酸构建模块在智能有机生物材料中应用的可能性. 类似地, Tao 等[44]在2019年使用色氨酸模拟FF的化学构象, 从而形成含苯环的二肽自组装纳米材料. 相对于FF, 该种纳米材料的化学结构内引入了更多氢键和苯环, 产生了较低的带隙, 能够在光刺激和热刺激下保持高稳定性, 兼具有显著的压电效应. 该晶体在黑暗环境中展现出约0.9 TΩ的高电阻, 但在紫外线照射下电阻降低至原有的1/3, 具备生物有机半导体光电应用潜质. 这些基础构建单元的拼接也能诞生新的自组织结构. 例如, Bera等[45]发现自然界中的脯氨酸和苯丙氨酸连接成的三肽(Pro-Phe-Phe), 不同于常见的二苯丙氨酸二肽β片组装结构, 能够自组装成独特的螺旋结构, 进而形成细长的螺旋纤维网络, 为生物分子纳米材料提供了超短螺旋三肽组件.
进一步地, 在已有的常见生物分子构建单元基础上, 人们从分子结构和分子手性角度探索生物分子的化学结构对组装结果的影响. 大量实验发现, 生物分子如多肽分子及其衍生物的氨基酸侧链基团、氨基酸手性对控制自组织过程的非共价相互作用力影响巨大. 生物分子的氨基酸侧链基团能够与水分子或其他侧链基团形成氢键, Yuan等[46]发现调整氨基酸侧链之间的氢键相互作用可以获得机械刚度、压电效应和热稳定性各有优势的生物晶体纳米材料. 如图1(b)所示, 分别用缬氨酸(Valine, Val)、亮氨酸(Leucine, Leu)和甲硫氨酸(Methionine, Met)制备了晶体, 尽管它们的侧链基团氢键排列相似, 但作用强度不同, 测试发现它们各自形成晶体的物理性质有所不同. 特别地, 高氢键强度的缬氨酸晶体具有最高的刚度, 氢键相互作用较弱的亮氨酸表现出低刚度和最强的压电效应. 这说明弱氢键相互作用会降低晶体的机械刚度, 有利于增大分子在应力下的位移以提高压电响应.
通常情况下, 天然蛋白质中的氨基酸为同手性, 而人工合成生物材料不存在氨基酸手性限制. Tao等[48]发现由色氨酸对映体拼接的L-色氨酸-D-色氨酸(L-tryptophan-D-tryptophan , Ww)简单二肽, 能够形成氨基酸晶体并在晶体内大量储存水分子. 这种氨基酸晶体在微观上呈现出二肽和水分子交替的“三明治”结构, 因而含有丰富的定向氢键, 保证了结构内部的电子跃迁, 最终实现压电效应提升. 同时, 作者通过碘吸附和同位素替换方法, 发现能够促进结构内部的电荷转移, 进一步提高了该材料的压电性能. Bera等[49]同样关注氨基酸手性对组装体结构和材料宏观性质的影响, 通过研究苯丙氨酸和色氨酸这两种氨基酸在不同手性组合下的二肽自组装性质, 验证了不同手性的同种氨基酸的自组装动力学存在的差异. 同种氨基酸对映体等摩尔混合后形成的外消旋混合物能够折叠组装成完美的“knob-to-hole”结构, 进而聚集组织成更大的薄片. 在提高晶体聚集倾向的同时, 有利于制造机械性能更出色的刚性晶体纳米材料. 随着氨基酸手性介导自组装纳米材料宏观性质研究逐渐深入, Ji等[47]探索了氨基酸对映体和非手性单元共聚形成的共晶中, 氨基酸的手性对晶体机械强度、压电响应等性质的影响. 乙酰化丙氨酸(acetylated alanine, AcA)在同手性下生长的晶体呈片状, 不同手性外消旋混合物生长的晶体呈树突状, 可将它们分别与非手性的1, 2-二(4-吡啶基)乙烯(1, 2-bis(4-pyridyl)ethylene, BPE)混合形成共晶. 按照图1(c)展示的组合方式, 对映体共组装共晶的杨氏模量更突出, 而单手性共晶光致发光和压电效应更加明显, 展现了基于氨基酸手性的微观差异在调控晶体宏观性质方面的应用潜能. 2022年, 在上述共晶材料的研究基础上, 我们将乙酰化谷氨酸(acetylated glutamic acid, AcE)和联吡啶(bipyridine, BPY)进行共组装[50]. 区别于BPY与水分子形成的共面分子排列, BPY与AcE在氢键作用下形成人字形分子排列, 促使晶体结构发生堆叠转变. 同时, BPY中的芳香基团能够在紫外激发光下发出蓝色荧光, AcE不含荧光基团, 而BPY/AcE共晶的晶体构象变化后, 荧光发射光谱发生红移呈现绿色荧光. 因此, 可综合使用不同的晶体构建块实现晶体宏观物理性质的多方面调控. 结合以上两个工作的研究成果, Ji等[51]在同年通过改变氨基酸手性方法, 将非手性的联吡啶衍生物1, 2-二(4-吡啶基)乙烷(1, 2-bis(4-pyridyl)ethane, BPA)与手性乙酰化丙氨酸进行共组装, 首次报道了中心对称结晶共晶体增加单晶压电响应特性的现象, 并实验总结了共结晶微观结构变化对宏观物理力学特性的影响.
在深刻理解生物分子化学结构调控自组织材料物理性质机制的基础上, 人们在已有的生物分子构建单元上接入改性组件, 以扩大其材料性能的优势. 如图2(a)所示, 2017年, Tao等[52]将5-单(4-羧基苯基)-10, 15, 20-三苯基卟啉(5-mono(4-carboxyphenyl)-10, 15, 20-triphenyl porphine, MCpP)与FF进行偶联, 得到了一种仿生构建模块MCpP-FF. 该模块能自组装成纳米纤维, 并聚集成多孔纳米微球. 纳米微球中的丰富芳香基团赋予其高度光敏感性和光电子传输能力, 可用作人工光合作用的光敏天线. 除此以外, 我们[53]又于2020年合作开发了基于核酸肽的生物构建块, 如图2(b)所示, 由二肽和二肽核酸自组装形成的超螺旋结构, 能在疏水作用下搭建成超分子框架结构; 超螺旋结构中的氢键一方面能够维持整个框架的机械结构稳定, 一方面能通过调节超螺旋手性改变框架空腔形态, 使组装体构成孔隙率可控的高机械刚度半导体光电材料. 进一步地, Yuan等[54]发掘了具有丰富氢键位点的含磺酸生物分子单元, 并将其作为高压电生物晶体的设计中心. 如图2(c)所示, 他们在牛磺酸分子(taurine)中引入酰胺基和环己基, 分别形成N-(2-乙酰氨基)-2-氨基乙磺酸(N-(2-Acetamido)-2-aminoethanesulfonic acid, ACES)和2-(环己胺)乙烷磺酸(2-(Cyclohexylamino) ethane sulfonic acid, CHES). 其中, ACES相对于牛磺酸的对称构象改变, 产生了更多氢键并形成类平行β片的堆叠结构, 呈现出高压电特性; 而CHES则维持中心对称结构, 堆积成类似反平行β片双层结构, 不具有压电特性. 该工作展示了将分子从中心对称转变成非中心对称构象的简单压电生物分子材料设计策略. 其次, 添加改性组件后的生物分子构建单元也能进行合理调整. Basavalingappa等[55]在2020年设计了含有更多芳香基的β, β-二苯基-丙氨酸-OH(β, β-diphenyl-Ala-OH, Dip)化学分子单元, 并用其取代线性丙氨酸二肽FF、环二苯基丙氨酸二肽(cyclo-FF)和叔丁氧羰基二苯基丙氨酸二肽(tert-butyloxycarbonyl diphenylalanine, Boc-FF)中的每个苯丙氨酸, 得到了全新的Dip-Dip, cyclo-Dip-Dip和Boc-Dip-Dip二肽. 作者期望更多的苯环能够增强π-π相互作用, 从而产生更高的压电效应和刚度. 三种苯环数量翻倍的超短二肽各自组装成不同的超分子结构, 均能生长成与FF类似的纳米棒结构. 此外, 二肽晶体内部苯环的紧密堆积还能产生与FF不同的层状组织结构, 呈现符合实验预期的更高刚度、压电性能和可见光波长范围内的光致发光. 如图2(d)所示, Dip-Dip从蓝光至红光区皆有荧光发射, 其杨氏模量和刚度与FF相比增加了数倍.
最后, 将发掘出的多种生物分子构建模块与功能化小分子系统结合, 能够实现自组装生物分子软物质纳米材料的应用开发. 例如, 将导电多肽纳米晶体与光催化金属离子系统结合, 用于人工光合作用方向的应用拓展. 本课题组[56]设计了一种不需要引入电介质的自组装多肽纳米管, 搭建出了能够实现光收集和催化的体系. 首先在自组装FF纳米管表面通过强疏水作用修饰钌配合物的配体, 使可吸收阳光产生激子的钌离子在FF纳米管表面进行配位, 进而在纳米管内产生光电子流, 之后在纳米管表面沉积铂纳米颗粒团簇接受能量流, 催化还原氧化型辅酶I, 成功将光能转化成化学能, 实现光合作用在多肽纳米管上的复现. 如前所述, Basavalingappa等[29,57]使用鸟嘌呤肽核酸(guanine peptide nucleic acid, G-PNA)与芴甲氧羰基(fluorenylmethyloxycarbonyl, Fmoc)缀合产物Fmoc-G-PNA自组装成的四聚体晶体形成纳米带, 与钌基光捕获天线和铂催化剂整合构成图3(a)中的光催化平台, 催化NAD+氧化型辅酶I还原为NAHD实现人工光合作用. 相比基于脱氧核糖核酸DNA的系统[58], 该工作采用的Fmoc-G-PNA在自组装过程中于分子头尾形成氢键产生环状四聚体结构, 四聚体间再垂直堆积组成类似四链体的稳定结构, 因此具有更好的机械稳定性、热稳定性和应用前景. 自组装生物分子纳米材料不仅广泛应用于宏观机电硬件材料的制备, 也可拓展至介观尺度的细胞层面. 本课题组[59]将贻贝粘附蛋白中的特殊氨基酸3, 4-二羟基苯丙氨酸(3, 4-dihydroxyphenylalanine, Dopa)修饰在能够在生理pH下自组装的可逆多肽末端上. 如图3(b)所示, 自组装形成的纳米纤维由于其Dopa的存在既可与细胞形成强粘附, 又能在pH的改变下随着可逆肽的解组装温和分离细胞, 成为可用于细胞捕获和释放的智能生物分子纳米纤维材料, 有望用于单细胞病理分析和细胞治疗. 除此以外, 我们还受到极地生物体内抗冻蛋白的启发, 模仿其三维结构设计了含有苏氨酸残基的自组装多肽[60]. 如图3(c)所示, 我们在一种常用的自组装多肽基序上分别增加一个或两个苏氨酸(Threonine, T)残基模仿抗冻蛋白, 又准备了添加天冬氨酸(Aspartic acid, D)的多肽作为对照组. 实验发现, 含苏氨酸的多肽确实抑制了冰晶生长, 并且苏氨酸越多, 抑制效果越好. 这表明多肽β片上的苏氨酸残基能够与冰晶排列匹配结合, 实现冰晶生长抑制. 这种可自组装的冰生长抑制肽作为新型生物冷冻保护剂, 有利于细胞、酶等的冷冻保存.
-
生物分子自组织形成的纤维网络在吸水膨胀后能够形成水凝胶, 从而构成自组织生物分子软物质凝胶材料. 其中, 多肽由于分子量较小、结构相较蛋白简单而易于设计, 具有良好的动态响应和生物相容性, 在生物分子凝胶材料领域具有独特优势, 非常适用于可降解的生物医用材料开发[61,62]. 为更好地了解自组装多肽软物质凝胶中多肽与凝胶性质之间的关系, Li与Cao[63]在2018年对多肽水凝胶的自组装物理化学机制进行了研究, 发现稳定分子间的弱相互作用力是多肽水凝胶制备的关键. 因此, 可以从氢键、疏水作用等相互作用力的角度对多肽进行设计和修饰, 或通过调节环境因素来控制多肽原纤维的生长或解聚, 使多肽自组装结构拥有更高的热力学稳定性和合适的动力学性能, 从而实现多肽水凝胶宏观理化性质的调控.
然而, 自组装多肽水凝胶较难克服多肽动态解离和聚合带来的凝胶力学强度较弱和结构不稳定等问题. 针对这一问题, 人们首先提出了引入共价交联位点以稳定整体凝胶网络的方法. 本课题组[64]在2013年报道了一种通过光交联法调控短肽水凝胶机械性能的方法. 如图4(a)所示, 我们首先将多肽预组装成纤维网络, 之后通过基于钌络合物的光催化方法使纤维上随机分布的酪氨酸残基二聚, 即在不稳定的物理缠结纤维网络中引入稳定的化学交联位点. 通过这种光交联方法, 可以在2 min内增强水凝胶的机械性能, 多肽水凝胶的机械性能提高104倍, 储能模量提升至约100 kPa. 更重要的是, 这种方法保留了多肽基生物材料的优良生物相容性, 大幅提升了水凝胶在水中的耐侵蚀能力和机械稳定性, 是目前报道的机械性质最优的小分子多肽水凝胶之一. 这种机械稳定性显著提升的光交联超分子水凝胶在组织工程和药物控制释放等领域具有广泛的应用前景. 另一方面, 通过调控环境因素使多肽组装成纤维网络过程中的非共价相互作用力增大或均衡, 也是稳定水凝胶的有效方法. 例如, Cheng 等[65]在2016年报道了一种通过酶调控多肽水凝胶pH实现凝胶解聚的方法. 作者认为, 酶诱导手段可以控制水凝胶形成后更接近平衡状态, 使用葡萄糖氧化酶和过氧化氢酶促进体系内葡萄糖酸的产生并释放质子, 温和下调体系内pH以促进多肽自组装, 从而改进水凝胶的机械性能, 这种方法有望成为构建血糖监测凝胶支架材料的新思路.
自组装多肽/蛋白水凝胶的低刚度和易降解特性在生物医学领域, 尤其是细胞生理研究中具有重要优势, 常用于模拟细胞外基质环境以研究细胞生理行为. 本课题组[66]设计了一种使用光响应蛋白Dronpa145N制备的水凝胶, 该水凝胶具有机械强度快速可逆并可时空调控的特点. 如图4(b)所示, 这种蛋白在405 nm光照下可形成四聚体, 而在505 nm光照下可恢复为单体状态, 从而使由蛋白与高分子聚合物形成的预聚物在凝胶和溶液状态之间切换, 其光响应动力学与体系中的蛋白浓度正相关. 该水凝胶作为硬度可调的细胞生长基底, 成功验证了细胞迁移率与基底硬度相关的普遍结果, 适合作为仿细胞外基质进行三维细胞培养, 并有望成为理想的细胞迁移模型系统, 帮助理解复杂的细胞功能. 进一步地, 适用于细胞培养的自组装多肽/蛋白水凝胶被广泛用于伤口愈合、软骨修复等方面的生物医学功能材料开发. 例如, Zhou等[67]采用了一种具有天然广谱抗菌活性的多肽自组装成水凝胶, 作为伤口敷料抑制细菌感染. 其中, 抗菌肽具有β折叠构象和极性与非极性氨基酸交替排列的化学结构, 能够在疏水作用驱动下组装成纳米纤维; 之后利用溶液环境中的磷酸基团形成氢键, 最终构成超分子水凝胶. 这种在类生理环境中形成的多肽水凝胶极大地保留了多肽的生物相容性和降解性, 表现出显著的抗菌和促进伤口愈合能力, 有潜力成为新型伤口敷料来提高烧伤和炎症创面的自修复速度.
小分子多肽因其序列较短, 相对蛋白质更易合成且成本较低, 使得基于多肽的自组织生物分子软物质凝胶材料在工业生产和生活应用方面具有良好的发展前景. Li与Wang[68]考虑到水凝胶的多孔结构在纺织工业中的染料废水处理方面具有理想的吸附特性, 且多肽通常含有的疏水基团与染料之间存在强烈的弱相互作用, 针对不同有机染料测试了光交联多肽水凝胶的染料吸附能力. 他们发现, 通过增强短肽与染料分子之间的静电相互作用, 可以提高水凝胶对染料的吸附能力, 从而启发了便捷、低成本的高效染料吸附凝胶的生产. 此外, 由于多种领域对能够在外部刺激下发生结构变化的水凝胶致动器有很高的需求, 本课题组[31]基于3, 4-二羟基苯丙氨酸(多巴, Dopa)的电化学响应特性构建了一种电控超分子水凝胶. 如图4(c)所示, 我们设计了含多巴的自组装肽, 当其多巴基团被电氧化还原时, 会发生多肽组装纤维可逆的形态和疏水性质转变, 从而引起超分子水凝胶的宏观体积和机械性能变化. 正如图4(c)中展示的, 罗丹明B和考马斯亮蓝两种染料分子分别混入水凝胶以模拟药物释放过程, 结果表明较长的氧化时间明显地延长了药物释放时间, 而还原态的水凝胶在较短的时间内释放了全部的染料分子. 这种电化学响应超分子水凝胶非常适合制作成电刺激转化机械能的致动器. 我们还展示了使用这种水凝胶构建的简单药物释放系统和微流控驱动装置, 预计可以在新型生物传感器、人造机器人等领域得到广泛应用.
-
由单一生物分子自组装的生物分子软物质材料, 因为其材料机制简单, 通常机械强度不是特别高, 功能应用范围有限. 为了拓展自组装生物分子软物质材料更多的应用场景, 人们使用多种材料构建方法制造复合材料[69]. 水凝胶双网络结构是水凝胶的常见有效增韧手段, 通常由一层紧密且易拉伸断裂的网络和一层松散但高韧性的网络组成. 紧密的网络用于能量耗散, 增强整体凝胶的韧性. 不同于传统双网络水凝胶, 本课题组[70]提出以聚丙烯酰胺高分子聚合物作为第一网络, 以在高应变下被破坏后能够快速重组装的短肽纤维作为第二网络, 成功制备了一种新型的聚合物-超分子聚合物双网络水凝胶. 在该水凝胶中, 短肽纤维作为凝胶网络中的可逆牺牲单元发挥着重要作用, 使双网络水凝胶的压缩杨氏模量最高可达1.6 MPa, 接近软骨的杨氏模量, 并具有类似软骨的应力松弛和快速恢复的力学性能. 在上述工作的双网络水凝胶设计基础上, 我们通过精细调整双重超分子网络的结构, 如图5(a)所示, 使多肽自组装成了刚度更高的纳米带[71], 而不是之前使用的纳米纤维. 这种纳米带结构降低了单个短肽的损伤以及超分子动态解离和重组对整体网络连续性的影响, 使最终得到的双网络水凝胶保持了出色的快速可逆恢复力学性能, 并且将杨氏模量和韧性显著提高了3—5倍.
固体纳米材料的引入是提高生物分子软物质材料复杂性和力学性能的另一有效方法. 本课题组[72]通过在自组装工程肽的凝胶预制溶液中混入二氧化硅纳米颗粒, 在自组装成凝胶后再加入酶催化多肽生成能与纳米颗粒交联的残基, 使纳米颗粒均匀固定在超分子网络中. 与直接将纳米颗粒分散进凝胶相比, 这种纳米颗粒交联多肽的凝胶稳定方法使机械强度提高了3000多倍. 石墨烯作为一种新型优异导电纳米材料, 常被引入生物分子软物质材料体系来优化结构和功能. 我们还报道了一种新型的石墨烯和多肽杂化可注射水凝胶[73]. 如图5(b)所示, 通过设计多肽一端可与氧化石墨烯在非共价相互作用下形成复合物, 一端可发生稳定的光交联, 从而形成含有石墨烯片层的凝胶网络; 而多肽与石墨烯之间的π–π堆积易在高应变等条件下被破坏, 而在低应变下保持高回复效率, 使水凝胶具有可注射特点, 有望应用于远程控制药物释放等领域. 除此以外, 我们[32]还提出一种包含多肽包覆石墨烯的聚丙烯酰胺水凝胶. 其中, 能够自组装成β片结构的多肽在C端连接聚乙二醇柔性长链, 在N端通过芘基在疏水作用和π-π堆积下覆盖在石墨烯表面, 并进一步构成多肽纤维网络. 与传统水凝胶电容器中凝胶作为导电层的“三明治”结构不同, 该工作将多肽和聚丙烯酰胺充当双层电介质, 石墨烯充当导电层, 构成了凝胶网络中的无数串并联微电容器. 自组装多肽网络与石墨烯之间存在稳定又动态的界面结合力, 有利于保持凝胶的自修复和高拉伸性能, 同时提高电容传感效率. 如图5(c)所示, 片状水凝胶材料在重复弯曲和拉伸循环下电信 号稳定, 弯曲时电容增加而拉伸时电容缩小; 柱状水凝胶材料在重复压缩循环下电信号稳定, 并且随着压力增大, 石墨烯复合层间距减小, 电容增大而电信号增强. 该材料还具有高可塑性, 支持3D打印成图5(c)中的复杂网络和多层支架结构. 总之, 该凝胶极大地提升了水凝胶人工皮肤传感器的耐拉伸、超灵敏和自修复性能, 为开发新型功能复杂的可穿戴设备提供了一个可行的新方案.
除了以上两种生物分子材料复合方式外, 还可以在自组织生物分子软物质材料中引入金属离子, 利用多肽-金属配位复合体赋予材料高机械强度、韧性和其他特殊性质和功能. 本课题组[74]通过引入金属配位复合物, 使透明的自组装多肽超分子水凝胶具有稳定均匀的荧光, 实现了不同荧光图案的3D打印. 我们设计的配体分子首先通过化学交联接入多肽水凝胶支架, 然后与金属离子配合提高凝胶网络结构的稳定性, 同时保证荧光发色基团分布均匀. 在此基础上, 切换水凝胶的配位过渡金属离子或调整合适的激发光, 即可实现水凝胶荧光颜色的改变. 在此基础上, 引入的多肽-金属离子配位存在多方面的可调控的影响因素, 为材料结构设计带来新的思路. Zeng等[75]关注金属配位键的动态交联性质, 通过调节凝胶内部每个短肽配体中金属离子结合位点的数量, 提高了凝胶网络的物理交联密度. 由于金属配位键分布较为均匀, 水凝胶的机械强度和韧性大大提高至1350 kJ/m3, 同时维持了原本由多肽组装体赋予的高拉伸和自修复性能, 可在伤口敷料、人造皮肤等生物医学领域得到拓展和应用. 在另外一个工作中, 本课题组[76]关注自然界中生物承重材料可否通过蛋白质弱相互作用的协同性来实现高机械稳定性和快速恢复能力, 设计了串联两个金属离子配位点的十肽, 并制备了水凝胶材料. 如图6(a)所示, 我们发现多配体的金属配位模式与单配体相比具有更强的结合力, 而双位点多配体的金属配位模式引入了过渡中间态, 使具有双结合位点的十肽在配位时发生协同作用, 提高了与金属离子的结合速率和结合强度. 这一设计成功地将水凝胶的断裂应力提升至大约3 MPa, 并保持了高水平的耐拉、防切割和快速回复性能, 有望成为软机器人制造中的承重组织构建单元. 值得注意的是, 一些具有特殊应用的金属离子与自组织生物分子软物质材料复合能够产生功能复杂的特殊材料. Wang等[77]利用可医用造影的钆离子作为交联剂, 与透明质酸-Dopa复合体中Dopa上的儿茶酚基团配位. 当钆离子浓度不断增加, 即交联点增加后, 能够形成机械强度随离子浓度升高增强的水凝胶. 在合适的钆离子浓度下, 该水凝胶具有剪切变稀特征, 适用于皮下注射输送药物至患处进行药物缓释, 完成如图6(b)所示的肿瘤内或皮下局部显影和疗效跟踪, 可作为原位疫苗接种治疗肿瘤的理想载体. 最后, 多肽-金属离子配位可与其他材料复合策略结合, 设计出微观结构更加有序的自组织生物分子软物质材料. 例如, 我们将添加柔性链隐藏长度的凝胶增韧策略与引入金属离子方法相结合, 构建了具有分级结构的超强超韧水凝胶[78]. 如图6(c)所示, 自组装多肽的一端通过丙烯酰胺单体聚合形成的长柔性链, 可在多肽排列组装后团缩成隐藏花边; 铜离子在多肽纤维间配位, 增强整个多肽复合体的机械稳定性; 复合体之间通过共价交联剂组合, 最终形成凝胶完整的分级结构. 这种水凝胶能在高应变下优先释放隐藏长度来提高韧性, 在高应力下发挥多肽和金属离子配位的协同作用保持高刚度、快速恢复和抗疲劳特性, 在仿生机器人组件制造方面具有广阔的应用发展潜力.
-
迄今为止, 人们对自然界生物分子各类自组织复杂结构已经有了广泛的了解, 解析的结构越来越多, 进而对其自组织结构形成的规律和其生物功能的发展也有了越来越深刻的认识. 毫无疑问, 这些对人工生物材料的设计和研发提供了重要的指导. 以多肽分子作为自组装模块或基元来构建生物分子软物质材料, 由于其生物分子的多样性和多功能性, 预计在未来的很长时间内将会有巨大的发展机遇. 值得注意的是, 基于多肽等生物分子的软物质材料不可避免地存在一些弱点, 如易被生物侵蚀、组装结构不稳定或不均匀、需要生理条件等. 这些弱点恰巧可在再生医学等需要可降解特性材料的领域[79]中得到合理的利用, 或者针对这些弱点, 探索开发出非共价和共价相互作用并存的各种结构来补偿和稳定[80].
受益于人工智能(artificial intelligence, AI)的发展[81–83], 天然进化的生物大分子和人工设计的小分子多肽分子的结构正以极快的速度被解构和分析[84,85]. 以自然为灵感选择生物体内的功能模仿对象, 人们可以通过AI辅助计算与设计, 设计出新的蛋白质组并对其成千上万的基序进行筛选[86], 通过分子动力学模拟等手段实施功能模拟验证, 避免大量枯燥冗余的重复性无效实验和实验材料资源浪费; 同时, 人们通过已经建立的生物分子数据库, 探索发展生物材料组学[87], 加速疾病和药物的高通量筛选[88]并不断建立新的生物医药数据库, 有利于提高生物软物质新材料的研发效率、增加创新性成果的产出和转化. 更重要的是, AI算法的迅猛发展极有希望促进自组织生物分子软物质材料设计理论的进步, 引导自组织生物分子软物质材料未来的持续性发展. 遗憾的是, 由于AI的各种模型算法均基于现有的天然蛋白质, 一方面, 还缺乏对复杂功能蛋白质物理原理的深刻理解; 另一方面, 目前的算法还不能生成超越天然蛋白质框架的全新蛋白质类型. 因此, AI辅助生物分子材料设计的算法仍亟待开发. 可以预见, 考虑到运用AI辅助带来的成本降低和样本量增大等优势, 使用人工智能深度学习蛋白机制, 筛选或设计有效蛋白基序, 模拟验证生物分子的功能和自组织过程, 进而模拟计算出生物分子材料构建的结构, 选出合理结构, 并投入实验生产测试. 这样的生物分子软物质材料设计流程, 可能是未来自组织生物分子软物质材料开发的主流趋势[89].
另一值得关注的自组织生物分子软物质材料的发展趋势, 是实现与尖端应用结合的产业转化, 将生物分子软物质材料真正应用于科技生活中, 拓展自组织生物分子软物质材料的创新应用范围和场景, 探索各类新型生物分子材料在不同领域可能的应用. 例如, 用于细胞疗法生产免疫细胞以治疗肿瘤等疾病的模块化平台和药物递送系统[90]、基于生物打印技术的人体仿生器官和软骨组织材料[23]、可制作成压电能量收集器、晶体管等的生物半导体元件[91–93]、生物基三维超导纳米结构[94], 具有可穿戴或可植入特性的生物柔性电子传感器[95,96]、神经假体和脑机接口[97]等方面的不断创新与发展. 发挥自组织生物分子软物质材料特有的快速动力学、高生物相容性等优势, 并结合AI技术, 将现有的自组织生物分子软物质材料整合集成为仿生软体机器人, 并使其能够自主运动和灵敏感知, 进而完成复杂的任务, 是生物分子材料领域最具吸引力的挑战.
总而言之, 当前自组装生物分子软物质材料领域非常需要学科之间的合作, 深入挖掘生物分子自组织的物理机制, 并运用生物、化学和材料等学科知识创新设计生物分子, 最后引入AI计算方法帮助最终成果在医学、电子等领域得到拓展和应用. 尽管因为生物分子的固有复杂性和不稳定性而充满挑战, 自组装生物分子软物质材料在多学科研究者们的共同努力下, 或将成为未来最有可能实现技术突破和产业化发展的领域之一.
自组装生物分子软物质材料及其物理特性
Self-assembled biomolecular soft materials and their physical properties
-
摘要: 自组装生物分子软物质材料是以生物分子或生物分子基元为构建单元, 通过自组织过程形成的一类新型软物质材料. 因其组成单元的生物特性和其中弱相互作用驱动组装的特征, 这类材料通常具有高度生物相容性、可逆组装、动态响应和微结构可控性等优势, 在生物医学、组织工程和柔性传感等领域中被广泛关注并得到了相关研发和应用. 本文简要介绍自组装生物分子软物质材料的基本构建原理和物理特性, 并以氨基酸、多肽分子等组装单元为例, 对三类自组装生物分子软物质材料(纳米材料、凝胶材料和复合材料)的自组装分子机制、材料构建思路、力学特性和功能应用场景做了具体阐述. 我们认为自组装生物分子软物质材料的研究, 将从结构单元的发掘和相关特性的表征, 向多功能性质定制与前端应用集成方向发展, 从而研发出崭新的复合智能生物软物质材料, 进一步促进其在生物医学、有机半导体和软体机器人等新兴领域中的应用.Abstract: Self-assembling biomolecular soft materials are a novel type of soft matter formed through the self-assembly process by using biomolecules or biomolecular building blocks. The characteristics of bio-sourced origin and assembly driven by weak interactions endow these materials with advantages such as high biocompatibility, reversible assembly, dynamic responsiveness, and controllable microstructures. These properties offer immense potential for development in fields such as biomedicine, tissue engineering, and flexible sensing. This paper concisely reviews the fundamental construction principles of self-assembling biomolecular soft materials and discusses three categories, i.e. nanomaterials, gel materials, and composite materials, by using amino acids and peptides as examples of assembly units. The specific self-assembly molecular mechanisms, material construction strategies, and functional application scenarios of these materials are elucidated. We anticipate that the research on self-assembling soft matter biomolecular materials will evolve from exploring structural units and measuring properties to customizing multifunctional properties and integrating advanced applications. This will lead to the development of novel composite intelligent biomolecular soft matter materials, and further promoting their applications in biomedicine, organic semiconductors, and soft robotics.
-
Key words:
- soft matter /
- biomolecule /
- self-assembling peptide /
- hydrogel /
- mechanical properties .
-

-
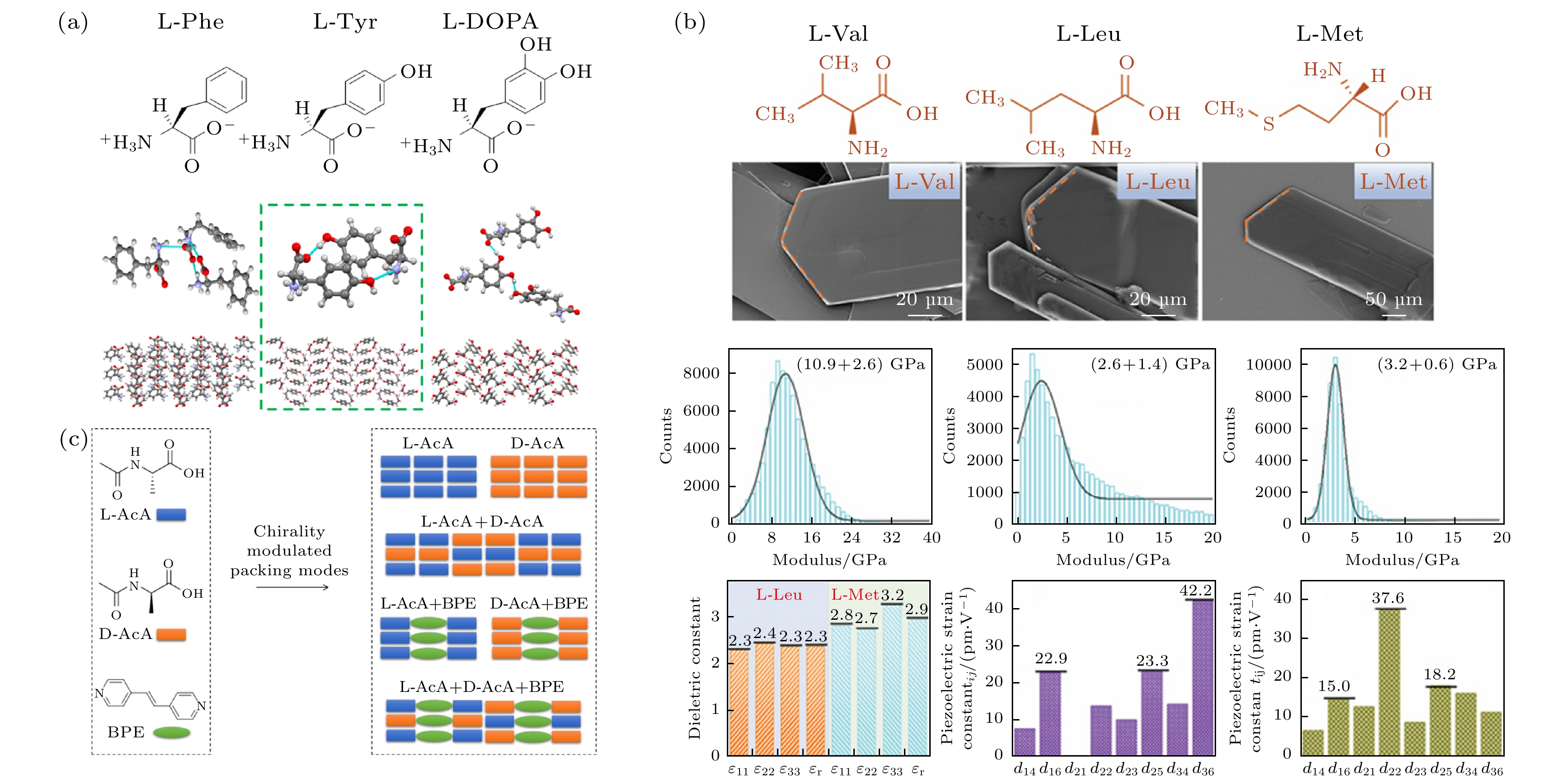
图 1 基于氨基酸分子的自组织生物分子软物质纳米材料的力学性质受堆叠方式、氢键和手性影响 (a) 苯丙氨酸(L-Phenylalanine, L-Phe)、酪氨酸(L-Tyrosine, L-Tyr)和多巴(L-DOPE)的化学结构和晶体结构和堆叠方式[43]; (b) 缬氨酸(Valine, Val)、亮氨酸(Leucine, Leu)和甲硫氨酸(Leucine, Met)的化学结构、扫描电子显微镜(scanning electron microscope, SEM)照片(上), 原子力显微镜(atomic force microscope, AFM)测定的杨氏模量(中)以及Leu和Met分别的介电常数和压电常数(下)[46]; (c) 调控1, 2-二(4-吡啶基)乙烯(1, 2-bis(4-pyridyl)ethylene, BPE)和不同手性的乙酰化丙氨酸(acetylated alanine, AcA)的晶体组装模式以制备宏观物理性质可调的晶体材料示意图[47]
Figure 1. The physical and mechanical properties of self-assembling biomolecular soft matter nanomaterials based on amino acids are affected by stacking mode, hydrogen bonding and chirality: (a) Chemical structures, crystal structures and packing of phenylalanine (L-Phe), tyrosine (L-Tyr) and dopa (L-DOPE)[43]; (b) chemical structures, scanning electron microscope (SEM) images (top) and Young’s modulus (middle) measured by atomic force microscope (AFM) of valine (Val), leucine (Leu), and methionine (Met); the calculated dielectric constants and piezoelectric constants of Leu and Met (bottom)[46]; (c) schematic diagram of regulating the crystal assembling models of 1, 2-bis(4-pyridyl)ethylene (BPE) and different chirality of acetylated alanine (either L-AcA or D-AcA) to prepare crystal materials with macroscopic tunable physical properties[47].
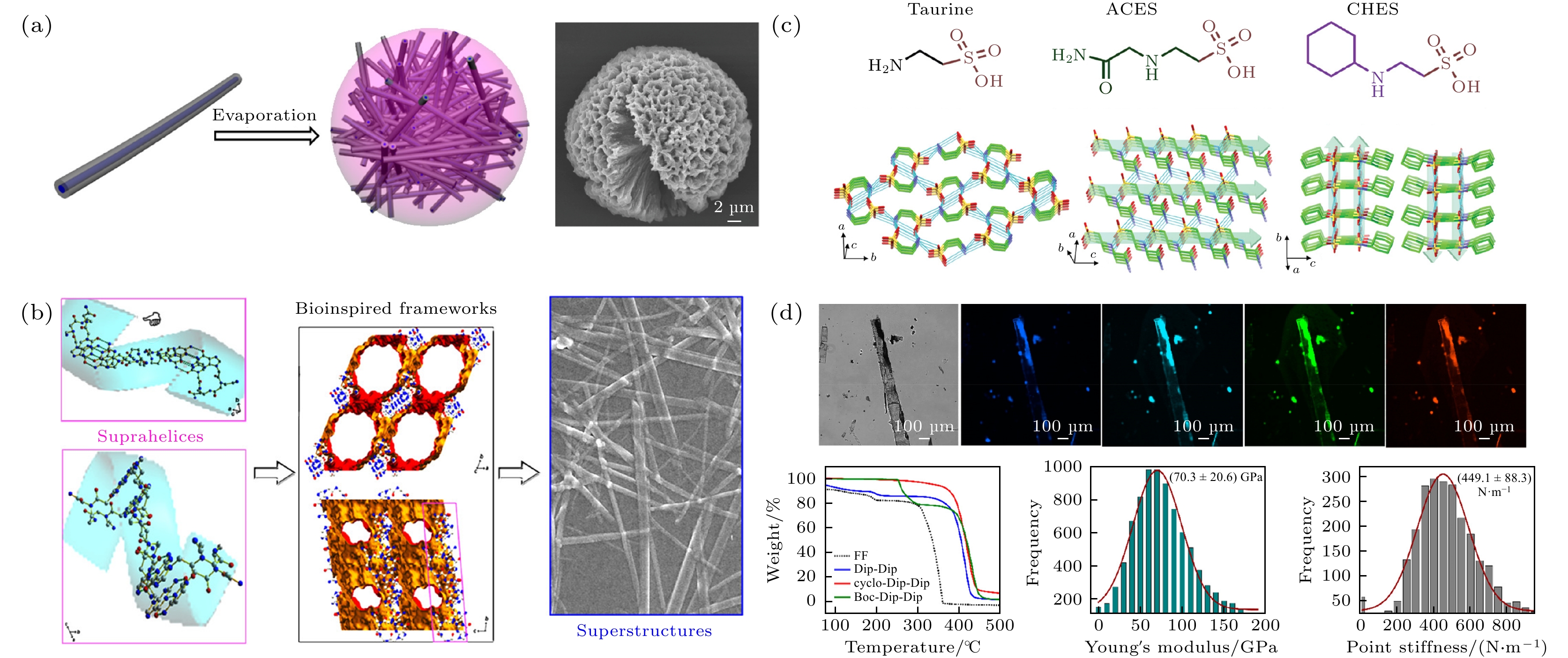
图 2 引入不同改性组件的自组织生物分子软物质纳米材料 (a) MCpP-FF纳米纤维在甲苯蒸发后聚集成的多空微球示意图及纳米微球的SEM照片[52]; (b) 二肽核酸自组装形成的超螺旋构象组成超分子框架的示意图和 SEM照片[53]; (c) 牛磺酸、ACES及CHES的化学结构(上)和超分子堆积示意图(下)[54]; (d) 含多苯环结构的短肽晶体的荧光照片(上), 热重分析和力学强度(下)[55]
Figure 2. Self-assembling biomolecular soft nanomaterials with different modified component: (a) Schematic diagram and SEM image of MCpP-FF nanofibers aggregated into porous microspheres after toluene evaporation[52]; (b) illustration and SEM image of supramolecular framework composed of the superhelix conformation formed by the self-assembly of dipeptide nucleic acid[53]; (c) chemical structures (top) and supramolecular packing diagram (bottom) of taurine, ACES and CHES[54]; (d) fluorescence images of short peptide crystals with polyphenyl structures (top), thermogravimetric analysis, and mechanical strength (bottom)[55].
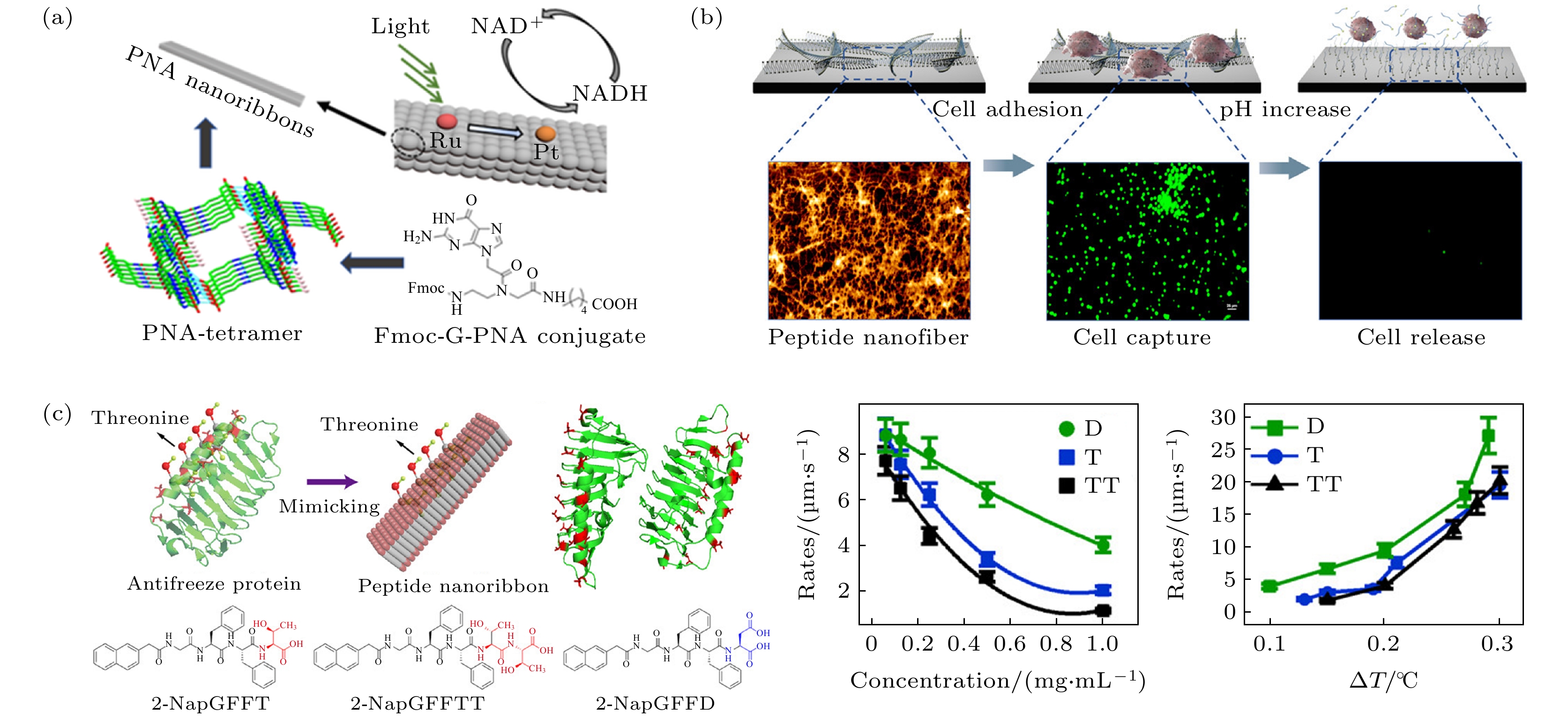
图 3 基于短肽及其衍生物的自组织生物分子软物质纳米材料应用开发 (a) 基于Fmoc-G-PNA缀合物的人工光合作用集成系统示意图[29]; (b) 含多巴的纳米纤维在pH调控下的细胞捕获和释放示意图(上)和光学照片(下)[59]; (c) 模仿抗冻蛋白结构的含苏氨酸的自组装多肽示意图(左上)、三种抑制肽的化学结构(左下)和随多肽浓度和过冷温度变化的冰晶生长速率(右)[60]
Figure 3. Applications of self-assembling biomolecular soft nanomaterials based on short peptides and their derivatives assemblies: (a) Schematic diagram of an integrated artificial photosynthesis system based on Fmoc-G-PNA conjugate[29]; (b) schematic diagram (top) and optical images (bottom) of cell capture and release of nanofibers containing Dopa under pH regulation[59]; (c) schematic diagram of threonine-containing self-assembling peptide mimicking the structure of antifreeze protein (upper left). Chemical structure of three peptides (lower left) and growth rate of ice crystals under different peptide concentration and supercooling temperature (right)[60].
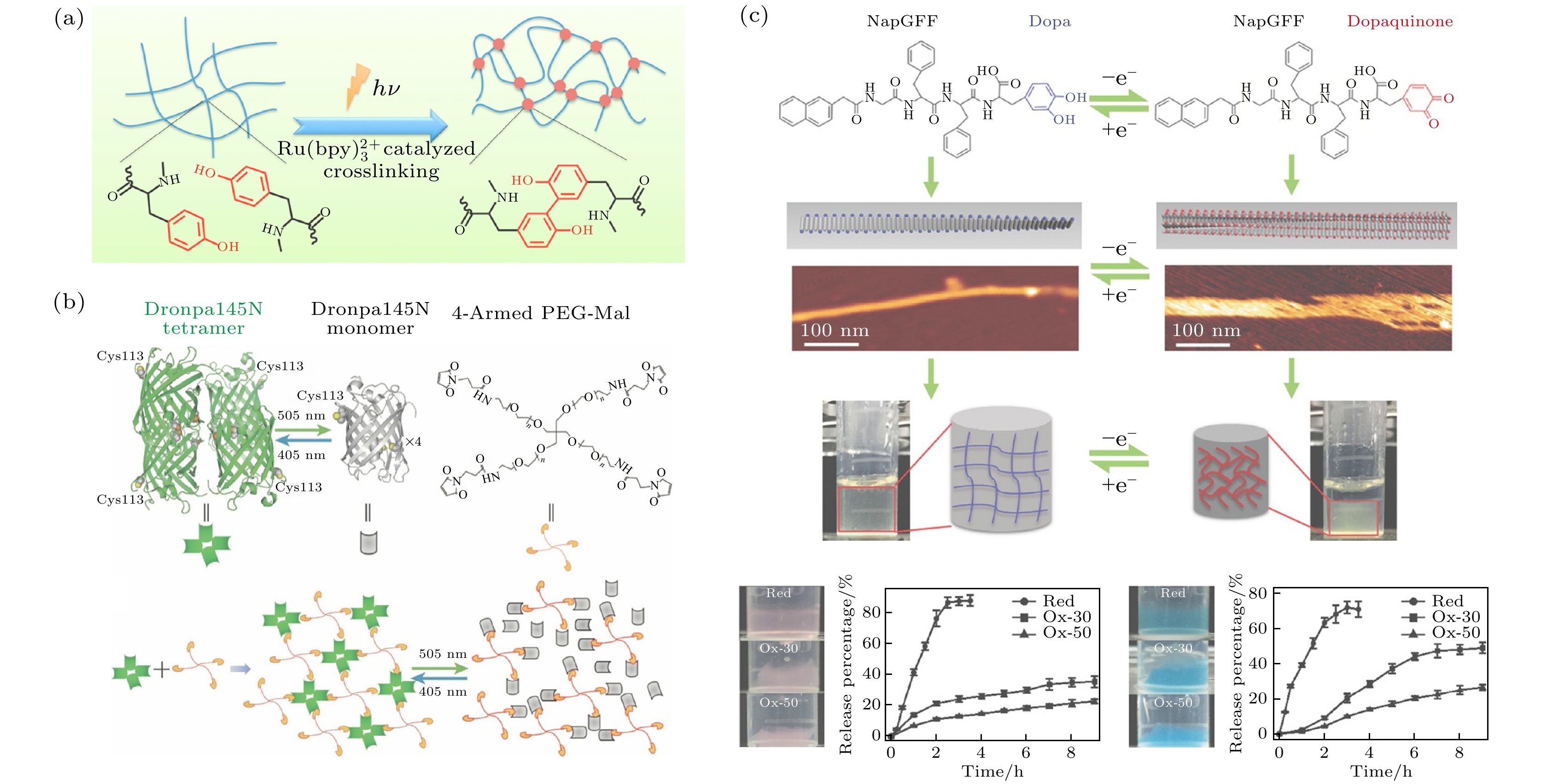
图 4 基于多肽及其衍生物的自组织生物分子水凝胶 (a) 钌络合物催化酪氨酸残基二聚的光交联方法增强凝胶机械稳定性示意图[64]; (b) 基于Dronpa145N的光响应水凝胶成胶原理示意图[66]; (c) 基于含多巴短肽可逆电氧化还原性质的超分子多肽水凝胶致动器设计(上)、电响应性质(中)和药物释放应用(下)[31]
Figure 4. Self-assembling biomolecular hydrogels based on peptides and their derivatives assemblies: (a) Schematic diagram of enhancing mechanical stability of hydrogel by photo-cross-linking strategy of tyrosine dimerization catalyzed by ruthenium complex[64]; (b) schematic diagram of photo-responsive hydrogels formation based on Dronpa145N[66]; (c) design (top), electro-response properties (middle), and drug release applications (bottom) of a supramolecular peptide hydrogel actuator based on the reversible electrochemical redox properties of dopa-containing short peptides[31].
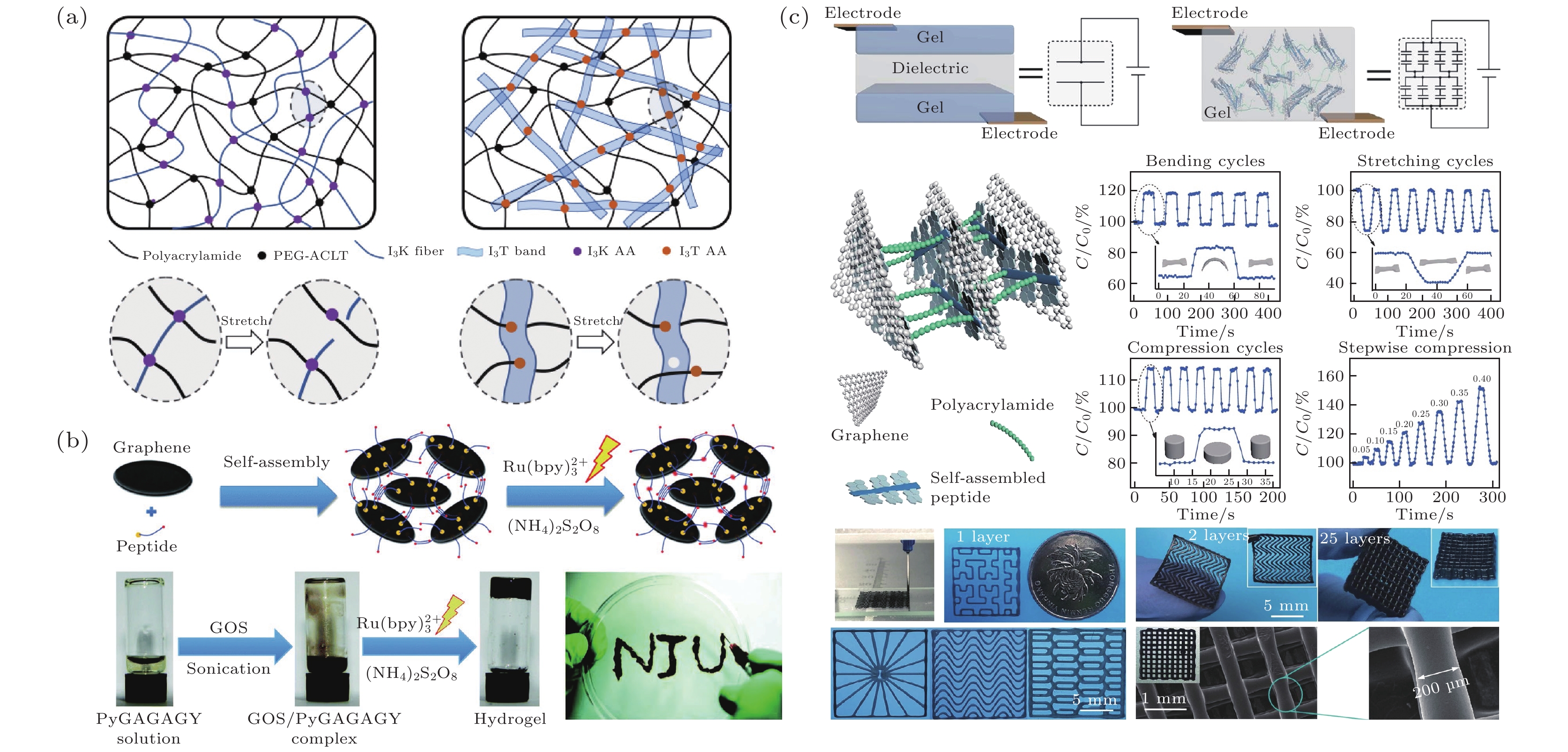
图 5 通过构建双网络或引入固体纳米材料构建自组织生物分子软物质复合材料 (a) 含自组装纳米纤维或纳米带的聚合物-超分子双网络水凝胶示意图[71]; (b) 石墨烯复合水凝胶的成胶原理示意图(上), 制备过程展示(左下)以及可注射性质展示(右下)[73]; (c) 传统“三明治”式凝胶和多肽包覆的石墨烯水凝胶的结构和等效电路示意图(上)、应变响应电容传感(中)和3D打印特性(下)[32]
Figure 5. Fabricating self-assembling biomolecular soft matter composite materials by constructing double networks or introducing solid nanomaterials: (a) Schematic diagram of polymer-supramolecular double network hydrogels containing self-assembled nanofibers or nanoribbons[71]; (b) schematic diagram of the gelation principle (top) and preparation process demonstration (lower left) of graphene hybrid hydrogel, as well as demonstration of its injectability (lower right)[73]; (c) structural and equivalent circuit diagram of traditional “sandwich” gel and peptide-coated graphene hydrogel (top), strain-responsive capacitance sensing (middle), and 3D printing characteristics (bottom)[32].
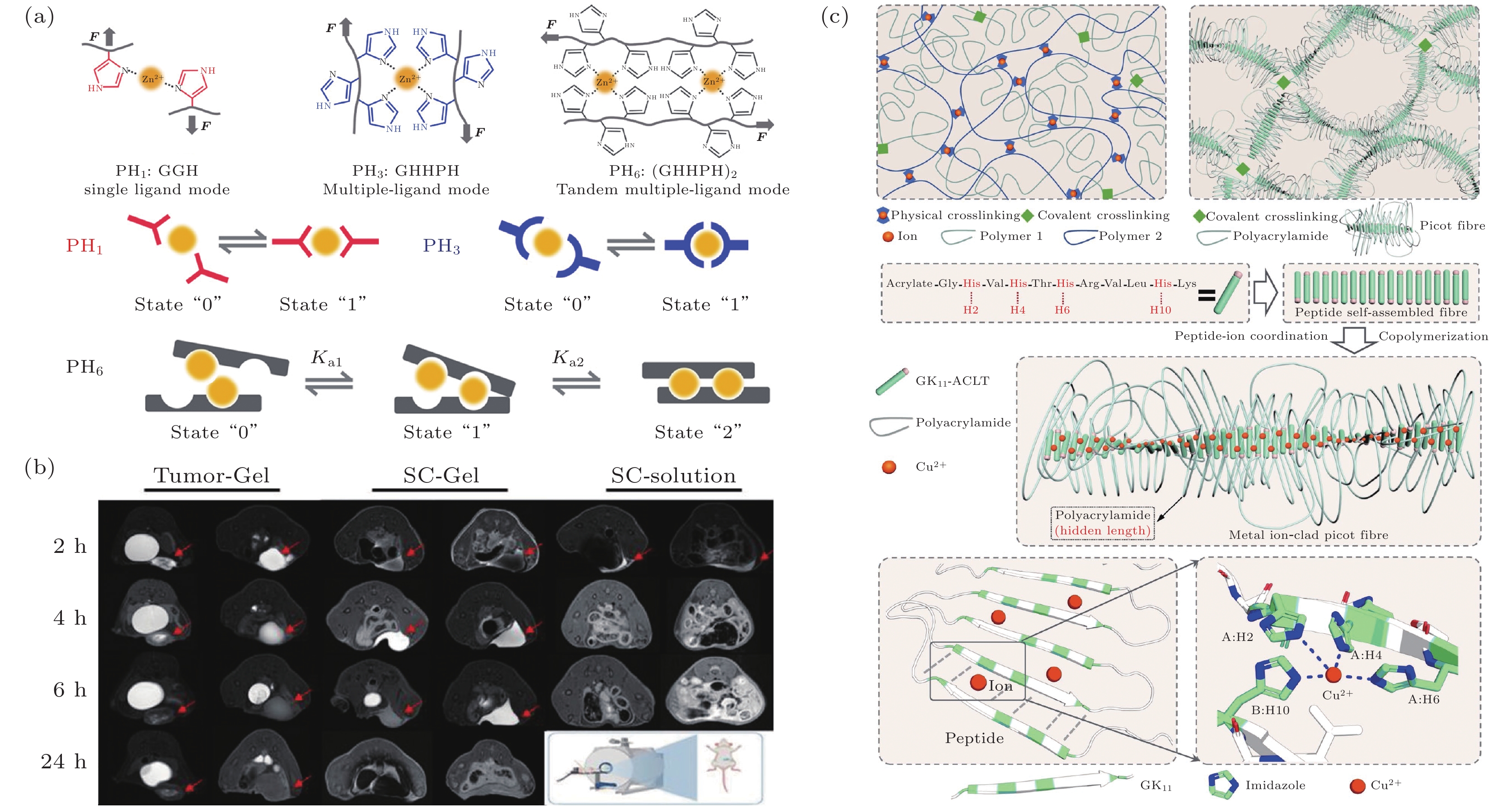
图 6 通过生物分子离子螯合构建自组织生物分子软物质复合材料 (a) 单结合位点的单配体和多配体金属离子配位, 以及双结合位点的多配体金属离子配位的分子机制和状态模型示意图[76]; (b) 体内肿瘤或皮下注射(SC)水凝胶和钆离子溶液的随时间推移的磁共振成像照片[77]; (c) 具有分级结构的强韧水凝胶构建示意图. 与传统含物理交联点的双网络水凝胶(左上)相比, 该水凝胶自组装多肽-配位铜离子-隐藏柔性链聚合物的分级结构既耗散能量又提升机械强度[78]
Figure 6. Fabricating self-assembling biomolecular soft matter composite materials through biomolecular-ion chelation: (a) Schematic diagram of molecular mechanism and state model of single ligand and multiple-ligand metal ion coordination with single binding sites and tandem multiple-ligand coordination with double binding sites[76]; (b) magnetic resonance imaging images over time of intratumor or subcutaneous injection (SC) by hydrogels or gadolinium ion solutions[77]; (c) schematic diagram of strong, tough hydrogel with hierarchical structure. Compared with traditional double network hydrogel by physical crosslinking (upper left), this hydrogel’s hierarchical structure of self-assembling peptide-coordination copper ion-hidden flexible polymer both dissipates energy and improves mechanical strength[78].
-
[1] Weitz D A 2022 Nat. Mater. 21 986 doi: 10.1038/s41563-022-01356-5 [2] Grzelczak M, Vermant J, Furst E M, Liz-Marzán L M 2010 ACS Nano 4 3591 doi: 10.1021/nn100869j [3] He M, Gales J P, Ducrot É, Gong Z, Yi G-R, Sacanna S, Pine D J 2020 Nature 585 524 doi: 10.1038/s41586-020-2718-6 [4] Kim Y J, Moon J B, Hwang H, Kim Y S, Yi G R 2023 Adv. Mater. 35 2203045 doi: 10.1002/adma.202203045 [5] Díaz-Villanueva J F, Díaz-Molina R, García-González V 2015 Int. J. Mol. Sci. 16 17193 doi: 10.3390/ijms160817193 [6] Shin Y, Brangwynne C P 2017 Science 357 eaaf4382 doi: 10.1126/science.aaf4382 [7] Soukasene S, Toft D J, Moyer T J, Lu H, Lee H K, Standley S M, Cryns V L, Stupp S I 2011 ACS Nano 5 9113 doi: 10.1021/nn203343z [8] Abbas M, Zou Q, Li S, Yan X 2017 Adv. Mater. 29 1605021 doi: 10.1002/adma.201605021 [9] Webber M J, Han X, Prasanna Murthy S N, Rajangam K, Stupp S I, Lomasney J W 2010 J. Tissue Eng. Regenerative Med. 4 600 doi: 10.1002/term.273 [10] Gu Z, Wang J, Fu Y, Pan H, He H, Gan Q, Liu C 2023 Adv. Funct. Mater. 33 2212561 doi: 10.1002/adfm.202212561 [11] Du X, Zhou J, Guvench O, Sangiorgi F O, Li X, Zhou N, Xu B 2014 Bioconjugate Chem. 25 1031 doi: 10.1021/bc500187m [12] Webber M J, Tongers J, Renault M A, Roncalli J G, Losordo D W, Stupp S I 2010 Acta Biomater. 6 3 doi: 10.1016/j.actbio.2009.07.031 [13] Webber M J, Appel E A, Meijer E W, Langer R 2016 Nat. Mater. 15 13 doi: 10.1038/nmat4474 [14] Levin A, Hakala T A, Schnaider L, Bernardes G J L, Gazit E, Knowles T P J 2020 Nat. Rev. Chem. 4 615 doi: 10.1038/s41570-020-0215-y [15] Parsons J T, Horwitz A R, Schwartz M A 2010 Nat. Rev. Mol. Cell Biol. 11 633 doi: 10.1038/nrm2957 [16] Theocharis A D, Skandalis S S, Gialeli C, Karamanos N K 2016 Adv. Drug Delivery Rev. 97 4 doi: 10.1016/j.addr.2015.11.001 [17] Hayes J D, Flanagan J U, Jowsey I R 2005 Ann. Rev. Pharmacol. Toxicol. 45 51 doi: 10.1146/annurev.pharmtox.45.120403.095857 [18] Xiang Y, Zhang J, Mao H, Yan Z, Wang X, Bao C, Zhu L 2021 Biomacromolecules 22 4846 doi: 10.1021/acs.biomac.1c01134 [19] Knowles T P J, Mezzenga R 2016 Adv. Mater. 28 6546 doi: 10.1002/adma.201505961 [20] Du X, Zhou J, Shi J, Xu B 2015 Chem. Rev. 115 13165 doi: 10.1021/acs.chemrev.5b00299 [21] Altunbas A, Lee S J, Rajasekaran S A, Schneider J P, Pochan D J 2011 Biomaterials 32 5906 doi: 10.1016/j.biomaterials.2011.04.069 [22] Hauser C A E, Zhang S 2010 Nature 468 516 doi: 10.1038/468516a [23] Zhang Y S, Yue K, Aleman J, Mollazadeh-Moghaddam K, Bakht S M, Yang J, Jia W, Dell’Erba V, Assawes P, Shin S R, Dokmeci M R, Oklu R, Khademhosseini A 2017 Ann. Biomed. Eng. 45 148 doi: 10.1007/s10439-016-1612-8 [24] Feiner R, Dvir T 2017 Nat. Rev. Mater. 3 17076 doi: 10.1038/natrevmats.2017.76 [25] Fu L, Li L, Bian Q, Xue B, Jin J, Li J, Cao Y, Jiang Q, Li H 2023 Nature 618 740 doi: 10.1038/s41586-023-06037-0 [26] Wang Y X, Geng Q, Lyu H, Sun W X P, Fan X Y, Ma K, Wu K, Wang J H, Wang Y C, Mei D Q, Guo C C, Xiu P, Pan D Y, Tao K 2024 Adv. Mater. 36 2401678 doi: 10.1002/adma.202401678 [27] Ren H, Xu T, Liang K, Li J, Fang Y, Li F, Chen Y, Zhang H, Li D, Tang Y, Wang Y, Song C, Wang H, Zhu B 2022 iScience 25 103673 doi: 10.1016/j.isci.2021.103673 [28] Rich S I, Wood R J, Majidi C 2018 Nat. Electron. 1 102 doi: 10.1038/s41928-018-0024-1 [29] Basavalingappa V, Xue B, Rencus-Lazar S, Wang W, Tao K, Cao Y, Gazit E 2020 ChemPhotoChem 4 5154 doi: 10.1002/cptc.202000083 [30] Willner I, Willner B 2001 Trends in Biotechnol. 19 222 doi: 10.1016/S0167-7799(01)01634-1 [31] Xue B, Qin M, Wang T, Wu J, Luo D, Jiang Q, Li Y, Cao Y, Wang W 2016 Adv. Funct. Mater. 26 9053 doi: 10.1002/adfm.201603947 [32] Xue B, Sheng H, Li Y, Li L, Di W, Xu Z, Ma L, Wang X, Jiang H, Qin M, Yan Z, Jiang Q, Liu J M, Wang W, Cao Y 2022 Natl. Sci. Rev. 9 nwab147 doi: 10.1093/nsr/nwab147 [33] Vale R D 2003 Cell 112 467 doi: 10.1016/S0092-8674(03)00111-9 [34] Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli R B G, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, Willbold D, Schröder G F 2017 Science 358 116 doi: 10.1126/science.aao2825 [35] Zhang S, Holmes T, Lockshin C, Rich A 1993 Proc. Nat. Acad. Sci. U. S. A. 90 3334 doi: 10.1073/pnas.90.8.3334 [36] Zhang S, Lockshin C, Cook R, Rich A 1994 Biopolymers 34 663 doi: 10.1002/bip.360340508 [37] Zhang S, Rich A 1997 Proc. Nat. Acad. Sci. U. S. A. 94 23 doi: 10.1073/pnas.94.1.23 [38] Caplan M R, Schwartzfarb E M, Zhang S, Kamm R D, Lauffenburger D A 2002 Biomaterials 23 219 doi: 10.1016/S0142-9612(01)00099-0 [39] Zhang S, Holmes T C, DiPersio C M, Hynes R O, Su X, Rich A 1995 Biomaterials 16 1385 doi: 10.1016/0142-9612(95)96874-Y [40] Davis M E, Motion J P M, Narmoneva D A, Takahashi T, Hakuno D, Kamm R D, Zhang S, Lee R T 2005 Circulation 111 442 doi: 10.1161/01.CIR.0000153847.47301.80 [41] Zou D, Cao Y, Qin M, Dai W, Wang W 2011 Chem. Commun. 47 7413 doi: 10.1039/c1cc12001h [42] Reches M, Gazit E 2003 Science 300 625 doi: 10.1126/science.1082387 [43] Ji W, Xue B, Arnon Z A, Yuan H, Bera S, Li Q, Zaguri D, Reynolds N P, Li H, Chen Y, Gilead S, Rencus-Lazar S, Li J, Yang R, Cao Y, Gazit E 2019 ACS Nano 13 14477 doi: 10.1021/acsnano.9b08217 [44] Tao K, Xue B, Li Q, Hu W, Shimon L J W, Makam P, Si M, Yan X, Zhang M, Cao Y, Yang R, Li J, Gazit E 2019 Mater. Today 30 10 doi: 10.1016/j.mattod.2019.04.002 [45] Bera S, Mondal S, Xue B, Shimon L J W, Cao Y, Gazit E 2019 Nat. Mater. 18 503 doi: 10.1038/s41563-019-0343-2 [46] Yuan H, Xue B, Yang D, Rencus-Lazar S, Cao Y, Gazit E, Tan D, Yang R 2023 Research 6 0046 doi: 10.34133/research.0046 [47] Ji W, Xue B, Bera S, Guerin S, Shimon L J W, Ma Q, Tofail S A M, Thompson D, Cao Y, Wang W, Gazit E 2021 Mater. Today 42 29 doi: 10.1016/j.mattod.2020.10.007 [48] Tao K, Donnell J O, Yuan H, Haq E U, Guerin S, Shimon L J W, Xue B, Silien C, Cao Y, Thompson D, Yang R, Tofail S A M, Gazit E 2020 Energy Environ. Sci. 13 96 doi: 10.1039/C9EE02875G [49] Bera S, Xue B, Rehak P, Jacoby G, Ji W, Shimon L J W, Beck R, Král P, Cao Y, Gazit E 2020 ACS Nano 14 1694 doi: 10.1021/acsnano.9b07307 [50] Ji W, Yuan H, Xue B, Guerin S, Li H, Zhang L, Liu Y, Shimon L J W, Si M, Cao Y, Wang W, Thompson D, Cai K, Yang R, Gazit E 2022 Angew. Chem. Int. Ed. 61 e202201234 doi: 10.1002/anie.202201234 [51] Ji W, Xue B, Yin Y, Guerin S, Wang Y, Zhang L, Cheng Y, Shimon L J W, Chen Y, Thompson D, Yang R, Cao Y, Wang W, Cai K, Gazit E 2022 J. Am. Chem. Soc. 144 18375 doi: 10.1021/jacs.2c06321 [52] Tao K, Xue B, Frere S, Slutsky I, Cao Y, Wang W, Gazit E 2017 Chem. Mater. 29 4454 doi: 10.1021/acs.chemmater.7b00966 [53] Tao K, Xue B, Han S, Aizen R, Shimon L J W, Xu Z, Cao Y, Mei D, Wang W, Gazit E 2020 ACS Appl. Mater. Interfaces 12 45192 doi: 10.1021/acsami.0c13295 [54] Yuan H, Cazade P-A, Zhou S, Shimon L J W, Yuan C, Tan D, Liu C, Fan W, Thangavel V, Cao Y, Thompson D, Yan X, Yang R, Xue B, Gazit E 2024 Small 20 2309493 doi: 10.1002/smll.202309493 [55] Basavalingappa V, Bera S, Xue B, O’Donnell J, Guerin S, Cazade P-A, Yuan H, Haq E u, Silien C, Tao K, Shimon L J W, Tofail S A M, Thompson D, Kolusheva S, Yang R, Cao Y, Gazit E 2020 ACS Nano 14 7025 doi: 10.1021/acsnano.0c01654 [56] Xue B, Li Y, Yang F, Zhang C, Qin M, Cao Y, Wang W 2014 Nanoscale 6 7832 doi: 10.1039/C4NR00295D [57] Basavalingappa V, Bera S, Xue B, Azuri I, Tang Y, Tao K, Shimon L J W, Sawaya M R, Kolusheva S, Eisenberg D S, Kronik L, Cao Y, Wei G, Gazit E 2019 Nat. Commun. 10 5256 doi: 10.1038/s41467-019-13250-x [58] Mutsamwira S, Ainscough E W, Partridge A C, Derrick P J, Filichev V V 2016 Chem. –Eur. J. ournal 22 10376 doi: 10.1002/chem.201601353 [59] Wang T, Li Y, Wang J, Xu Y, Chen Y, Lu Z, Wang W, Xue B, Li Y, Cao Y 2020 ACS Biomater. Sci. Eng. 6 6800 doi: 10.1021/acsbiomaterials.0c01485 [60] Xue B, Zhao L, Qin X, Qin M, Lai J, Huang W, Lei H, Wang J, Wang W, Li Y, Cao Y 2019 ACS Macro Lett. 8 1383 doi: 10.1021/acsmacrolett.9b00610 [61] Anderson S B, Lin C C, Kuntzler D V, Anseth K S 2011 Biomaterials 32 3564 doi: 10.1016/j.biomaterials.2011.01.064 [62] Sridhar B V, Brock J L, Silver J S, Leight J L, Randolph M A, Anseth K S 2015 Adv. Healthcare Mater. 4 702 doi: 10.1002/adhm.201400695 [63] Li Y, Cao Y 2018 Chin. J. Polym. Sci. 36 366 doi: 10.1007/s10118-018-2099-6 [64] Ding Y, Li Y, Qin M, Cao Y, Wang W 2013 Langmuir 29 13299 doi: 10.1021/la4029639 [65] Cheng W, Li Y 2016 Sci. Chin. -Phys. Mech. Astron. 59 678711 doi: 10.1007/s11433-016-0083-4 [66] Wu X, Huang W, Wu W H, Xue B, Xiang D, Li Y, Qin M, Sun F, Wang W, Zhang W B, Cao Y 2018 Nano Res. 11 5556 doi: 10.1007/s12274-017-1890-y [67] Zhou J, Cha R, Wu Z, Zhang C, He Y, Zhang H, Liu K, Fareed M S, Wang Z, Yang C, Zhang Y, Yan W, Wang K 2023 Nano Today 49 101801 doi: 10.1016/j.nantod.2023.101801 [68] Li Y, Wang L 2016 Chem. Lett. 45 1253 doi: 10.1246/cl.160597 [69] Goor O J G M, Hendrikse S I S, Dankers P Y W, Meijer E W 2017 Chem. Soc. Rev. 46 6621 doi: 10.1039/C7CS00564D [70] Sun W, Xue B, Li Y, Qin M, Wu J, Lu K, Wu J, Cao Y, Jiang Q, Wang W 2016 Adv. Funct. Mater. 26 9044 doi: 10.1002/adfm.201603512 [71] Wang T, Zhang Y, Gu Z, Cheng W, Lei H, Qin M, Xue B, Wang W, Cao Y 2021 Chin. J. Chem. 39 2711 doi: 10.1002/cjoc.202100370 [72] Li Y, Ding Y, Qin M, Cao Y, Wang W 2013 Chem. Commun. 49 8653 doi: 10.1039/c3cc45127e [73] Wu J, Chen A, Qin M, Huang R, Zhang G, Xue B, Wei J, Li Y, Cao Y, Wang W 2015 Nanoscale 7 1655 doi: 10.1039/C4NR05798H [74] Xia Y, Xue B, Qin M, Cao Y, Li Y, Wang W 2017 Sci. Rep. 7 9691 doi: 10.1038/s41598-017-10162-y [75] Zeng L, Song M, Gu J, Xu Z, Xue B, Li Y, Cao Y 2019 Biomimetics 4 36 doi: 10.3390/biomimetics4020036 [76] Sun W, Xue B, Fan Q, Tao R, Wang C, Wang X, Li Y, Qin M, Wang W, Chen B, Cao Y 2020 Sci. Adv. 6 eaaz9531 doi: 10.1126/sciadv.aaz9531 [77] Wang C, Jing Y, Yu W, Gu J, Wei Z, Chen A, Yen Y T, He X, Cen L, Chen A, Song X, Wu Y, Yu L, Tao G, Liu B, Wang S, Xue B, Li R 2023 Adv. Healthcare Mater. 12 2300877 doi: 10.1002/adhm.202300877 [78] Xue B, Bashir Z, Guo Y, Yu W, Sun W, Li Y, Zhang Y, Qin M, Wang W, Cao Y 2023 Nat. Commun. 14 2583 doi: 10.1038/s41467-023-38280-4 [79] Pina S, Oliveira J M, Reis R L 2015 Adv. Mater. 27 1143 doi: 10.1002/adma.201403354 [80] Xia D, Wang P, Ji X, Khashab N M, Sessler J L, Huang F 2020 Chem. Rev. 120 6070 doi: 10.1021/acs.chemrev.9b00839 [81] Thorn A 2022 Curr. Opin. Struct. Biol. 74 102368 doi: 10.1016/j.sbi.2022.102368 [82] Dai X, Chen Y 2023 Adv. Mater. 35 2204798 doi: 10.1002/adma.202204798 [83] Elofsson A 2023 Curr. Opin. Struct. Biol. 80 102594 doi: 10.1016/j.sbi.2023.102594 [84] Szymczak P, Szczurek E 2023 Curr. Opin. Struct. Biol. 83 102733 doi: 10.1016/j.sbi.2023.102733 [85] Wu X, Lin H, Bai R, Duan H 2024 Eur. J. Med. Chem. 268 116262 doi: 10.1016/j.ejmech.2024.116262 [86] Kortemme T 2024 Cell 187 526 doi: 10.1016/j.cell.2023.12.028 [87] Basu B, Gowtham N H, Xiao Y, Kalidindi S R, Leong K W 2022 Acta Biomater. 143 1 doi: 10.1016/j.actbio.2022.02.027 [88] Parvatikar P P, Patil S, Khaparkhuntikar K, Patil S, Singh P K, Sahana R, Kulkarni R V, Raghu A V 2023 Antiviral Res. 220 105740 doi: 10.1016/j.antiviral.2023.105740 [89] King N P, Bale J B, Sheffler W, McNamara D E, Gonen S, Gonen T, Yeates T O, Baker D 2014 Nature 510 103 doi: 10.1038/nature13404 [90] Eckman N, Nejatfard A, Cavet R, Grosskopf A K, Appel E A 2024 Nat. Rev. Bioeng. 2 408 doi: 10.1038/s44222-023-00148-z [91] Tao K, Makam P, Aizen R, Gazit E 2017 Science 358 eaam9756 doi: 10.1126/science.aam9756 [92] Zhang L, Lu J R, Waigh T A 2021 Adv. Colloid Interface Sci. 287 102319 doi: 10.1016/j.cis.2020.102319 [93] Zhao J W, Liu Q X, Tong X Y, Wang Y H, Cai K Y, Ji W 2024 Adv. Funct. Mater. 34 2401466 doi: 10.1002/adfm.202401466 [94] Shani L, Michelson A N, Minevich B, Fleger Y, Stern M, Shaulov A, Yeshurun Y, Gang O 2020 Nat. Commun. 11 5697 doi: 10.1038/s41467-020-19439-9 [95] Cheng X, Shen Z, Zhang Y 2023 Nat. Sci. Rev. 11 nwad314 doi: 10.1093/nsr/nwad314 [96] Nair V, Dalrymple A N, Yu Z, Balakrishnan G, Bettinger C J, Weber D J, Yang K, Robinson J T 2023 Science 382 eabn4732 doi: 10.1126/science.abn4732 [97] Tang X, Shen H, Zhao S, Li N, Liu J 2023 Nat. Electron. 6 109 doi: 10.1038/s41928-022-00913-9 -
计量
- 文章访问数: 742
- HTML全文浏览数: 742
- PDF下载数: 16
- 施引文献: 0