
 首页
首页 登录
登录 注册
注册



-
氨气作为一种极易溶于水且具有强烈刺激气味的有毒气体, 广泛存在于工业生产和医疗等领域, 对人们日常生活造成了一定的负面影响[1,2]. 研究表明, 长时间接触高浓度的氨气会刺激人体的呼吸系统、眼睛和皮肤, 对健康造成危害[3,4]. 此外, 氨气也是大气污染源之一, 能够与空气中的氮化物和硫化物反应, 生成硫酸铵和硝酸铵. 因此, 实时监测氨气浓度显得尤为重要. 在过去几十年间, 氨气传感器得到了快速发展, 涌现出多种以金属氧化物、金属硫化物和碳纳米管为气敏材料的氨气传感器[5–8]. 石墨烯因其超大的比表面积、高载流子迁移率、良好的强度和韧性, 以及优异的导电性能和光学特性而备受关注[9–11]. 2007年, Schedin等[12]利用氧化硅晶体表面的石墨微机械裂解技术制备出基于石墨烯的氨气传感器. 然而, 石墨烯的完整结构和零带隙特性限制了其气敏特性. 因此, 研究人员开始通过对石墨烯进行掺杂, 或将氧化石墨烯(graphene oxide, GO)作为气敏材料, 以开发对氨气具有响应特性的气体传感器. 据报道, Peng与Li[13]利用第一性原理(DFT)研究了氨气在石墨烯和氧化石墨烯表面的最佳吸附位置. 此外, Luo等[14]从理论上研究了碱土金属(Be, Mg, Ca和Sr)掺杂石墨烯对氨气的吸附能和灵敏度的影响. Park等[15]和Raza等[16]则分别通过对氧化石墨烯掺杂氮(N)和氟(F)原子, 实现了对氨气的检测. Tran等[17]采用溶液法合成技术制备了rGO/AgNWs混合纳米结构, 并成功实现了在室温下对氨气的检测. 接下来, Karaduman等[18]通过对比Ag, Au和Pd的特性, 发现Ag修饰的还原氧化石墨烯展现出了良好的气敏特性. 与传统的电极式有源气体传感器不同, Zhang等[19]利用近场电磁耦合技术构建了一种基于NFC的无线无源的Ag-rGO复合结构氨气传感器. 近年来, 一些学者开始研究Ag或Cu与石墨烯复合结构的气体传感器, 但对于Ag或Cu与石墨烯复合结构的氨气传感器的理论计算和传感器响应增强原理的研究仍然较为有限[20–22].
本研究利用第一性原理(DFT)方法计算了体系的电荷布居、态密度和吸附能, 探讨了GO, Ag掺杂氧化石墨烯(AgGO)与Cu掺杂氧化石墨烯(CuGO)对NH3的吸附机理. 首先, 针对不同含氧基团(羟基、羧基和环氧基)的GO对NH3的吸附特性进行了研究. 然后, 通过Ag或Cu原子的掺杂调控GO对NH3的吸附性能. 本研究从吸附距离、电荷转移、吸附能和态密度等方面分析了NH3在GO表面的吸附机理, 为进一步开发性能优异的氨气传感器提供了理论基础.
-
石墨烯模型采用包含32个C原子的4×4超级晶胞进行建模, 石墨烯平面与XY平面平行. 同时, 在Z方向设置了30 Å (1 Å = 10–10 m)的真空层, 以减少相邻石墨烯层之间的相互作用. 布里渊区积分使用4×4×1中心的Monkhorst-Pack (MP)网格进行结构优化, 并使用4×4×1中心的MP网格进行态密度(DOS)计算. 计算得到的石墨烯晶格常数为2.46 Å. 几何优化和相关电子参数采用Materials Studio 2020的DMol3模块进行第一性原理计算. 在DMol3模块中, 对于电子密度变化较大的体系, 局域密度近似(LDA)可能导致较大误差. 相反, 对于电子密度变化较小的体系, 广义梯度近似(GGA)通常能提供更准确的能量和结构计算[23]. 本文采用GGA和Perdew-Burke-Ernzerhof (PBE)泛函描述相互作用关系, 并选择DFT-D的Grimme方法对范德瓦耳斯力进行校正. 所有原子自旋不受限制, 采用全电子核心处理和具有偏振的双数值基组(DNP), 该基组与6-31G*基组相当[24,25]. 布里渊区的k点设置为4×4×1, 采用0.005 Ha的费米弥散和6.1 Å的全局轨道截止. 在几何优化方面, 能量收敛标准设置为1.0×10–6 Ha; 最大力容差设置为0.002 Ha/Å; 最大位移公差设置为0.005 Å. 在能量计算中, 自洽场容差设置为1.0×10–6 Ha/atom. 在几何优化过程中, 晶格参数不变.
氧化石墨烯模型的建立是通过在石墨烯模型上引入含氧基团实现的. Lerf等[26]和Szabó等[27]提出的Lerf-Klinowski模型和Szabo-Dekany模型被认为是最接近氧化石墨烯结构和性质的模型, 认为氧化石墨烯中主要存在环氧基、羟基和羧基这三种含氧基团. 本文在石墨烯表面引入羟基和环氧基, 在石墨烯边缘引入羧基, 形成三种带有不同含氧基团的氧化石墨烯模型, 并展开了对这三种含氧基团的研究. 基于上述氧化石墨烯模型, 将表面的一个C原子替换为Ag或Cu原子, 得到了Ag或Cu掺杂的氧化石墨烯模型, 其原子掺杂比例为3.13%. 图1展示了这些模型优化前的结构. NH3分子在所有模型中的吸附位置均设定在距表面3 Å处. 在图1(a)中, NH3与石墨烯表面平行, 置于碳原子上方; 图1(b)—(d)中, NH3与GO表面平行, 并放置于与含氧基团相连的碳原子上方; 图1(e)—(j)中, NH3与GO表面平行, 并放置于包含Ag或Cu以及含氧基团的碳六圆环中心位置. NH3分子在GO或XGO (X表示Ag或Cu)的吸附能计算如下:
其中
$ {E}_{{\mathrm{G}}{\mathrm{O}}/X{\mathrm{G}}{\mathrm{O}}+{{\mathrm{N}}{\mathrm{H}}}_{3}} $ 为GO或XGO吸附一个NH3分子的总能量;$ {E}_{{\mathrm{G}}{\mathrm{O}}/X{\mathrm{G}}{\mathrm{O}}} $ 为GO或XGO的总能量;$ {E}_{{{\mathrm{N}}{\mathrm{H}}}_{3}} $ 为NH3分子的总能量. 研究Ag和Cu原子掺杂GO体系的气敏性能, 需要探讨掺杂GO体系的稳定性. 因此, 本文计算了系统掺杂后的结合能. 结合能的计算如下:其中Eb为结合能; EXGO为XGO的总能量; EGV为含单空位GO的能量; EX为X金属原子的能量. 计算得到AgGO-O, AgGO-OH和AgGO-COOH的结合能分别为–2.49 eV, –1.94 eV和–2.36 eV, CuGO-O, CuGO-OH和CuGO-COOH的结合能分别为–4.47 eV, –4.24 eV和–4.91 eV. 6种掺杂GO的结合能均为负值, 说明Ag和Cu原子掺杂GO体系具有良好的稳定性.
-
为了研究GO中不同含氧基团对NH3的吸附机理, 本文分别研究了环氧基、羟基和羧基对NH3的气敏特性, 并将其与石墨烯进行了比较. 吸附NH3分子后的模型分别记为GO-O-NH3, GO-OH-NH3, GO-COOH-NH3和G-NH3. 图2为含有不同含氧基团的GO表面吸附NH3后的几何构型图, 而表1总结了各模型优化后的键长、键角和吸附距离. 环氧基和羟基位于GO表面, 而羧基位于GO边缘, 这与Anton等[26]提出的Lerf-Klinowski模型一致.
从几何构型的角度看, 与表面碳原子相比, 图2(b)—(d)中的三种含氧基团对NH3分子具有更强的捕获能力, 体现为吸附距离更近的趋势, 这可能是因为GO表面的含氧基团为NH3提供了更多吸附位点. 在吸附作用方面, 气体分子与被吸附表面的距离在一定程度上反映了吸附作用的强度. 吸附距离越小, 则吸附作用越强; 反之, 吸附作用越弱或者不存在. 根据表1所列, G-NH3, GO-O-NH3, GO-OH-NH3和GO-COOH-NH3的吸附距离分别为3.24 Å, 2.28 Å, 1.80 Å和2.44 Å. 吸附距离的大小顺序为G-NH3 > GO-COOH-NH3 > GO-O-NH3 > GO-OH-NH3. 在三种含氧基团中, GO-OH-NH3的吸附距离最小(1.80 Å), 表明GO中的羟基对NH3的吸附作用较强. 相比之下, 石墨烯对NH3的吸附距离最大(3.24 Å), 显示出石墨烯对NH3的吸附能力较弱. 因此, 这也说明在石墨烯中引入含氧基团可以一定程度上提高对NH3的吸附能力.
通过计算电荷总密度、差分电荷密度和Mulliken布居, 进一步分析石墨烯与GO对NH3吸附作用的详细机理. 图3展示了NH3分子、石墨烯和GO形成的电荷密度区域. 电荷密度区域的重叠情况反映了对气体分子的吸附特性. 若电荷密度区重叠, 说明存在较强的化学吸附; 反之, 若电荷密度区未重叠, 则表明吸附作用较弱, 存在较弱的物理吸附. 观察到图3中GO-OH-NH3模型, NH3分子与GO中的羟基之间存在明显的电荷密度重叠, 表明GO-OH-NH3发生了较强的化学吸附; 而G-NH3, GO-O-NH3和GO-COOH-NH3之间没有电荷密度重叠, 显示出较弱的物理吸附. 化学吸附相对物理吸附更强, 这一特性通过表1的吸附距离数据分析得到了验证.
图4显示了石墨烯和GO吸附NH3时的差分电荷密度图, 不同颜色区域显示了电荷的分布趋势, 蓝色表示电子积累区, 黄色表示电子耗尽区. 在图4(c)中, 可以清晰地观察到黄色区域围绕着羟基中的H原子, 而蓝色区域围绕着NH3分子中的N原子. 差分电荷密度图显示, GO表面H原子周围的电荷耗尽区几乎与NH3分子中N原子周围的电荷积累区相接触, 表明电荷从H原子周围的电荷耗尽区转移到N原子的电荷积累区. 根据表2中的Mulliken布居分析结果, 吸附前NH3分子的电荷量为0, 吸附后NH3分子的电荷变化量表示从石墨烯或GO向NH3分子转移的电荷量. 从表2数据可以得出, 在G-NH3, GO-O-NH3, GO-OH-NH3和GO-COOH-NH3中, NH3分子的电荷变化量分别为0.022e, 0.005e, 0.078e和0.036e. 这些电荷变化量为正, 表明在吸附过程中NH3从石墨烯或GO获得了电子. 尤其在GO-OH-NH3中, NH3分子的电荷变化量达到最高值为0.078e, 较高的电荷转移量通常意味着更强的相互作用, 结合差分电荷密度图, 可以得出GO-OH中的羟基与NH3分子之间存在较强的相互作用.
根据表2显示, 吸附能均为负值, 表明该过程为放热反应, 而放热反应则有利于吸附过程的进行. GO中不同含氧基团对NH3分子的吸附能绝对值大小排序为羟基(0.60 eV)>羧基(0.17 eV)>环氧基团(0.11 eV), 该结果与Peng和Li[13]的研究结论一致. 吸附能的绝对值越大, 表明系统结构越稳定, 吸附作用也越强. 结合表1中三种含氧基团对NH3的吸附距离, 可以推断三种含氧基团对NH3的吸附能力由强到弱依次为羟基、羧基和环氧基团.
图5展示了G和GO的能带结构, 其中E = 0 eV为费米能级. 由图5(a)可见, G的带隙宽度为0 eV, 导带底和价带顶在布里渊区中心相接. 结合图6(a)的情况, 可以得出G为零带隙半导体材料, 这与Liu等[28]的研究结果一致. 此外, G的零带隙能带结构容易受外部电场、表面吸附和晶格掺杂等因素的影响, 可能导致载流子类型的转变, 从而引起带隙打开现象[29,30]. 当引入含氧基团形成GO后, 与G相比, GO的能带结构呈现明显差异. 如图5(b)—(d)所示, 费米能级附近的能带曲线出现明显的曲率减小区域. 在图5(b)中, C原子和O原子的成键导致狄拉克点左移, 使得费米能级附近的态密度(DOS)更为陡峭[31,32]. 这也导致产生了0.101 eV的小带隙, 带隙增加意味着电子从导带跃迁到价带所需能量增加, 系统的电导率下降, 阻抗增大. 因此, 吸附NH3后, GO-O与NH3之间的电荷转移量降低, 使得GO-O-NH3的吸附能相比G-NH3减小. 在图5(c)和图5(d)中, 狄拉克点被移除, 费米能级上出现具有小峰的能带, DOS在费米能级附近也发生了显著的变化(图6(c)和图6(d)). 这可能与羟基和羧基修饰石墨烯表面有助于促进基底与NH3分子之间的电荷转移有关.
图6展示不同结构吸附NH3后的态密度. 带有不同含氧基团的GO吸附NH3后, 与G吸附NH3相比, 对应的DOS呈现出不同程度的变化. 图6(c)和图6(d)显示, 带有羟基和羧基的GO的DOS在费米能级附近均产生了显著变化, 而图6(b)显示, 带有环氧基的GO的DOS在费米能级附近变化不大. 通过对比可知, 羟基的引入导致在费米能级附近的变化明显大于羧基和环氧基.GO-OH-NH3具有最大的吸附能和Mulliken转移电荷, 表明羧基与NH3分子的相互作用比其他含氧基团更强, 更有利于GO对NH3分子的吸附. 在–20 eV时, 图6(c)和图6(d)均出现了明显的特征峰, 这可能与羟基和羧基提供了更多的吸附位点有关. 更多的吸附位点有利于对NH3的吸附, 因此相对于环氧基和原始石墨烯, 羟基和羧基对NH3具有更强的吸附能力.
综合考虑吸附距离、电荷密度、差分电荷密度、Mulliken转移电荷、吸附能以及态密度等因素, 对带有三种含氧基团的GO以及原始石墨烯对NH3分子的吸附机理进行了分析. 在吸附过程中, NH3分子充当电子受体, 而G和GO则充当电子供体, 电子由G和GO转移到NH3中. 电子转移量的大小反映了吸附强度. 研究结果显示, 在GO对NH3的吸附过程中, 羟基表现出较强的吸附能力, 羧基次之, 而环氧基团的吸附能力最弱.
-
对带有不同含氧基团的GO进行Ag掺杂, 并系统地研究了掺杂前后其对NH3分子的吸附机理. 建立了相关掺杂模型, 通过将GO-O-NH3, GO-OH-NH3和GO-COOH-NH3表面同一位置的C原子替换为Ag原子, 分别得到了AgGO-O-NH3, AgGO-OH-NH3和AgGO-COOH-NH3模型. 图7展示了经过几何优化后的GO和AgGO吸附NH3的俯视图, 图7(a)—(c)与图7(d)—(f)分别为Ag掺杂前后GO的俯视图. 通过比较发现, 图7(a)—(c)中, 所有NH3分子倾向于靠近含氧基团, 说明掺杂前的GO对NH3的吸附能主要源于含氧基团; 而在图7(d)—(f)中, 所有NH3分子在吸附过程中从含氧基团的中心位置向Ag原子靠近, 表明AgGO中的Ag原子对NH3分子具有更强的吸附作用.
表3总结了关于AgGO的吸附特性所需的相关参数, 包括吸附距离、电荷转移和吸附能. 分析吸附距离发现, Ag掺杂后, NH3分子倾向于与AgGO中的Ag原子接触, 而不是靠近含氧基团. 具体而言, 几组AgGO吸附NH3时, N原子与Ag原子的吸附距离均为2.29 Å, 表明Ag原子在吸附过程中扮演主导角色, 展现出比含氧基团更强的吸附作用. 在观察电荷转移与吸附能之间的关系时, 发现NH3分子中更多的电荷转移并不意味着系统具有更高的吸附能. 例如, 在表3中, AgGO-O-NH3中NH3分子的电荷转移量(0.171e)最大, 但其吸附能为–1.25 eV, 低于AgGO-COOH-NH3的吸附能(–1.39 eV). 此外, AgGO-COOH-NH3中Ag原子的电荷转移量为–0.082e, 明显高于AgGO-O-NH3中Ag原子的电荷转移量(–0.038e). 虽然AgGO-O-NH3和AgGO-COOH-NH3中NH3分子的电荷转移量几乎相同(分别为0.171e和0.170e), 但AgGO-COOH-NH3的吸附能(–1.39 eV)明显高于AgGO-O-NH3的吸附能(–1.25 eV). 这表明在吸附过程中, Ag原子比含氧基团具有更强的作用力. 在该情况下, Ag充当电子供体, NH3作为电子受体, 导致大量电荷从Ag原子转移到NH3分子. 在AgGO-OH-NH3中, Ag原子的电荷转移量(–0.033e)低于AgGO-O-NH3(–0.038e), 而AgGO-OH-NH3的吸附能(–1.26 eV)则略高于AgGO-O-NH3(–1.25 eV). 表明在Ag原子的电荷转移量相近的情况下, 含氧基团对系统的吸附能产生显著影响. 由前文可知, 羟基的吸附能力明显优于环氧基, 这在一定程度上解释了AgGO-OH-NH3的吸附能略高于AgGO-O-NH3的原因. 综上所述, 可以得出结论, AgGO的吸附能受含氧基团和Ag原子的协同效应影响. 从表3可以看出, Ag掺杂能显著地提高系统的吸附能. 进行Ag掺杂后, GO-O-NH3和GO-COOH-NH3的吸附能均提高了一个数量级, 而GO-OH-NH3的吸附能则提高了约2倍. 这可能是因为Ag具有比含氧基团更强的吸附作用, 而Ag的掺杂导致表面产生缺陷, 引入更多的吸附位点, 有利于气体的吸附作用.
图8(a)—(c)展示了Ag掺杂后系统的DOS变化, 在费米能级两侧均可以观察到明显的DOS变化, 表明Ag掺杂有助于提高系统对NH3的吸附性能. 特别是图8(c)中, 费米能级两侧的变化更为显著, 表明Ag掺杂的AgGO-COOH能显著增强对NH3分子的吸附能力, 与表3中吸附能提高的结果一致. 图8(d)—(f)和图8(g)—(i)研究了Ag原子和NH3分子中的N原子的部分态密度(PDOS), 有助于更好地理解NH3分子与AgGO的相互作用. 根据图8(d)—(f)和图8(g)—(i)中显示的信息, 可以观察到Ag原子和N原子的s, p和d轨道之间的共振现象, 显示出Ag原子和N原子之间形成了化学键. 这也证实了Ag原子与N原子之间存在较强的相互作用, 从而解释了Ag掺杂后系统吸附能增强的原因.
通过比较掺杂前后的GO, 发现在吸附NH3分子时, Ag原子与N原子之间存在较强的相互作用. 在掺杂后的系统中, Ag原子的吸附作用明显优于GO中的含氧基团, 导致吸附能发生显著变化. 从DOS和PDOS的角度解释了产生这种变化的原因: Ag原子在吸附过程中与N原子形成了化学键, 使系统结构更加稳定. 此外, 在更复杂的系统中, 不能仅根据电荷转移量来判断吸附性能的优劣, 因为吸附能的大小受多种因素共同影响.
-
将AgGO模型中的Ag原子替换为Cu原子, 获得Cu掺杂的GO相关模型, 分别命名为CuGO-O-NH3, CuGO-OH-NH3以及CuGO-COOH-NH3. 带有不同含氧基团的AgGO和CuGO吸附NH3时的电荷总密度图如图9所示. 从图9(a)—(f)中可以观察到, NH3分子与各个AgGO或CuGO间均存在显著的电荷密度重叠, 表明Ag或Cu原子在AgGO或CuGO中对NH3分子表现出较强的化学吸附.
表4总结了AgGO和CuGO吸附NH3的电荷转移和吸附能数据. 数据显示, 相较于AgGO-O-NH3 (0.171e), CuGO-O-NH3 (0.192e)中NH3分子的电荷转移提高了0.021e; CuGO-OH-NH3(0.180e)比AgGO-OH-NH3 (0.160e)中NH3分子的电荷转移提高了0.020e; CuGO-COOH-NH3 (0.192e)相比AgGO-COOH-NH3 (0.170e), NH3分子的电荷转移增加了0.022e. 综合分析发现, 相对于Ag掺杂系统, Cu掺杂系统中NH3分子的电荷转移量均增加了约0.020e, 表明NH3在CuGO中吸收了更多电子, 从而增强了其对NH3的吸附作用. 特别是在带有羟基和环氧基的GO中进行Cu掺杂时, 展现出比Ag掺杂更出色的吸附效果, 对应的吸附能分别增强了0.17 eV和0.14 eV; 而对于带羧基的GO, 吸附能减少了0.05 eV. 在AgGO中, 不同含氧基团对NH3的吸附能排列顺序为—COOH> —OH > —O—; 而在CuGO中, 吸附能排序为—OH> —O—> —COOH; 而在GO中, 排序为—OH> —COOH> —O—. 这说明仅通过含氧基团对NH3的吸附能力来评估金属掺杂的GO的吸附能大小是不全面的. 系统变得更为复杂, 吸附能的大小受系统中的含氧基团和金属原子共同作用的影响.
根据掺杂金属的类型及其与碳原子的距离, 可以将掺杂分为p型掺杂和n型掺杂, 这可能导致狄拉克点发生偏移[33,34]. 如图10所示, AgGO和CuGO的能带结构显示, Ag或Cu掺杂后, 狄拉克点向左移动. 具体而言, 在图10(a), (d)中, AgGO-O和CuGO-O的带隙为0 eV, 可能是由于C原子和O原子成键所致. 而在图10(b), (c)中, AgGO-OH和AgGO-COOH的带隙分别为0.391 eV和0.353 eV; 在图10(e), (f)中, CuGO-OH和CuGO-COOH的带隙分别为0.520 eV和0.183 eV. 带隙的开启会增加载流子浓度, 加强电荷转移, 同时增加系统对NH3分子的吸附能. 图11展示了AgGO或CuGO吸附NH3的系统中Ag或Cu与NH3的DOS图. 当空间中两个原子距离较近且两者的DOS在某些能量区间有交叠时, 可以推断这两个相邻原子之间存在化学键. 如图11(a)—(f)所示, Ag或Cu原子的DOS均与NH3的DOS之间存在交叠能量区间, 表明Ag或Cu与NH3之间形成化学键, 进一步证实了图9中电荷密度重叠区域存在化学吸附的现象.
-
通过深入研究带有不同含氧基团的GO及其Ag与Cu掺杂对NH3分子的吸附特性, 本文系统地分析了GO对NH3的吸附机理. 通过综合电荷总密度、差分电荷密度、电荷转移、吸附能和态密度等方面的研究, 深入地揭示了吸附过程的机制. 研究结果显示, 在GO吸附NH3的过程中, 羟基呈现明显的化学吸附特性, 而其他两种基团则表现为物理吸附, 其中羟基的吸附能力最强, 羧基次之, 环氧基最弱. 同时, 通过Ag或Cu原子掺杂调控GO对NH3的吸附作用, 电荷密度重叠以及Ag或Cu原子与N原子的轨道共振表明了其对NH3为化学吸附, 且掺杂后GO对NH3的吸附性有显著提升. 相较而言, CuGO对NH3表现出更强的吸附能力. 值得注意的是, 吸附NH3能力的强弱受金属原子和含氧基团共同作用影响. 研究结果表明, Ag或Cu原子掺杂能显著提高GO对NH3的吸附能力.
Ag, Cu掺杂氧化石墨烯吸附NH3的第一性原理研究
First-principles study of NH3 adsorption on Ag- and Cu doped graphene oxide
-
摘要: 基于第一性原理的密度泛函理论, 系统地研究了氧化石墨烯及其Ag与Cu掺杂对NH3分子的吸附特性. 通过计算电荷分布、态密度、能带结构和吸附能, 研究了含氧基团及金属掺杂对氧化石墨烯气敏性能的调控效应. 通过对掺杂Ag原子的氧化石墨烯的态密度进行分析, 发现Ag原子与NH3中的N原子的s, p和d轨道之间存在共振, 表明Ag原子和N原子之间形成了化学键. 这种化学键导致Ag对NH3的吸附作用明显强于含氧基团, 从而使得掺杂后的氧化石墨烯的吸附能提升了数倍. 此外, Cu掺杂氧化石墨烯同样能够显著地提升其对NH3的吸附性能. 在掺杂浓度均为3.13%的条件下, Cu掺杂的氧化石墨烯对NH3表现出更强的吸附能力. 同时, Ag或Cu掺杂的氧化石墨烯中, 羧基和环氧基对NH3的吸附模式由物理吸附转变为化学吸附; 而羟基则在掺杂前后始终显示出化学吸附特性. 综上所述, 金属掺杂的氧化石墨烯的吸附能会受到含氧基团和金属原子共同作用影响, 且Ag或Cu原子掺杂能显著提高氧化石墨烯对NH3的吸附性能.Abstract: Graphene has attracted great attention due to its large specific surface area, high charge carrier mobility, and excellent electrical conductivity. However, the inherent structural integrity and zero bandgap characteristics of graphene limit its gas sensing properties. Consequently, researchers have embarked on exploring avenues such as doping graphene or using graphene oxide as a gas-sensitive material to design gas sensors that respond optimally to ammonia. This work, based on first-principle density functional theory, focuses on the field of ammonia gas sensors, investigating in detail the adsorption characteristics of ammonia molecules on graphene oxide (GO) and graphene oxide doped with Ag and Cu (AgGO, CuGO). By calculating parameters including charge distribution, density of states, band structures, and adsorption energy, this work delves into the influences of diverse oxygen-containing groups and metal doping on the gas sensing properties of graphene oxide. The research results show that there is a substantial charge density overlap between the density of states of hydroxyl groups in graphene oxide and NH3 molecules, indicating a clear tendency towards chemical adsorption. It is particularly noteworthy that after NH3 adsorption, the graphene oxide containing hydroxyl shows the highest charge transfer (0.078e) and adsorption energy (0.60 eV), which indicates that the adsorption efficacy of NH3 is higher, followed by carboxyl groups and epoxy groups, which mainly participate in physical adsorption. Furthermore, this work delves into the influence of metal doping on graphene oxide, demonstrating that the adsorption capability of doped graphene oxide hinges upon the synergistic influence of oxygen-containing groups and metal atoms, with Ag-doped graphene oxide showing a several-fold increase in adsorption energy. Through the analysis of density of states, it is found that Ag atoms resonate with s, p, and d orbitals of the N atom in NH3, proving the formation of a chemical bond between Ag atom and N atom. Moreover, a comparative analysis shows that Cu-doped graphene oxide (CuGO) has an increased charge transfer of about 0.020e and slightly higher adsorption energy than Ag-doped graphene oxide (AgGO) when adsorbing NH3. Intriguingly, under the same doping concentration, CuGO exhibits superior adsorption performance to NH3. It is worth noting that in graphene oxide doped with Ag or Cu, the adsorption mechanism of carboxyl and epoxy groups transforms from physical adsorption into chemical adsorption, while the hydroxyl groups maintain consistent chemical adsorption properties before and after doping. This indicates that doping with Ag or Cu atoms can significantly enhance the adsorption capability of graphene oxide to NH3.
-
Key words:
- doping /
- graphene oxide /
- first principle /
- adsorption .
-

-
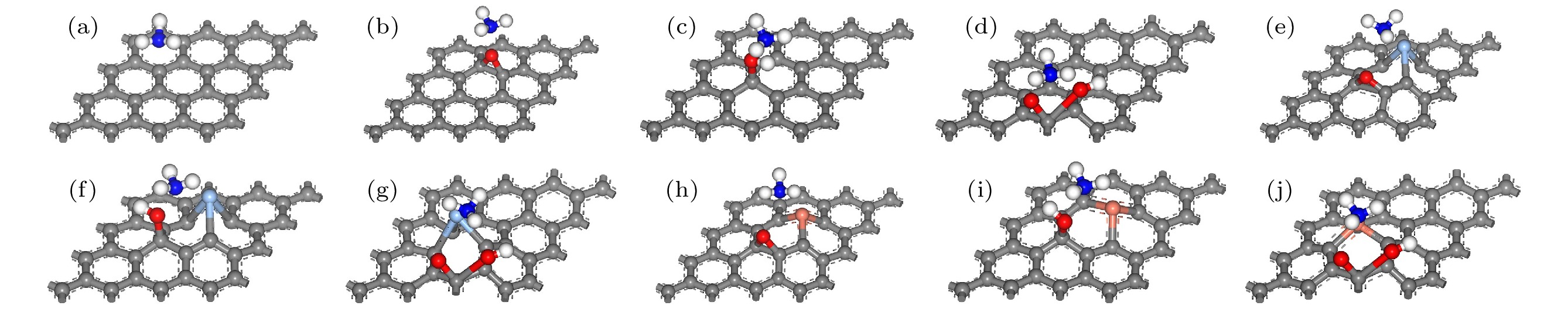
图 1 优化前相关建模图 (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3; (e) AgGO-O-NH3; (f) AgGO-OH-NH3; (g) AgGO-COOH-NH3; (h) CuGO-O-NH3; (i) CuGO-OH-NH3; (j) CuGO-COOH-NH3
Figure 1. Related model diagrams before optimization: (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3; (e) AgGO-O-NH3; (f) AgGO-OH-NH3; (g) AgGO-COOH-NH3; (h) CuGO-O-NH3; (i) CuGO-OH-NH3; (j) CuGO-COOH-NH3.
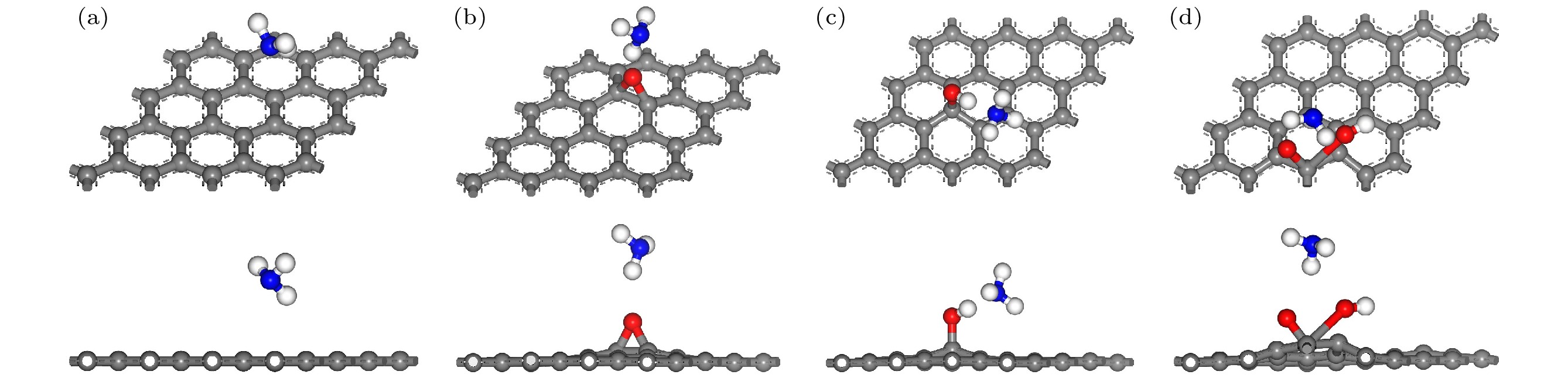
图 2 优化后G和含有不同含氧基团的GO吸附NH3的俯视图和侧视图 (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3
Figure 2. Top and side views of optimized G and GO with different oxygen-containing groups adsorbing NH3: (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3.
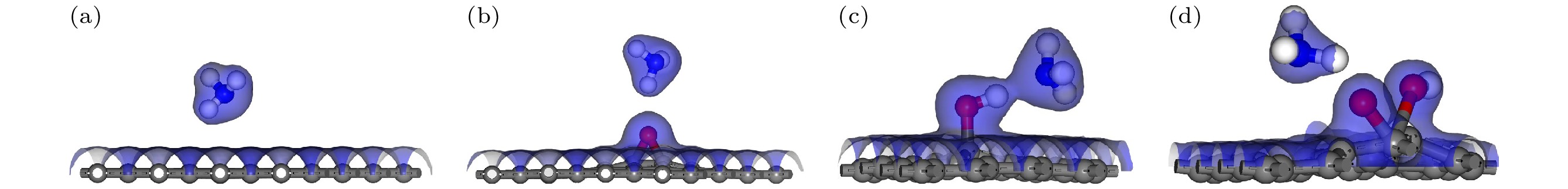
图 3 G和含有不同含氧基团的GO吸附NH3的电荷总密度图(等值面为0.2) (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3
Figure 3. Total charge density of G and GO with different oxygen-containing groups adsorbing NH3 (The isosurface value is 0.2): (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3.
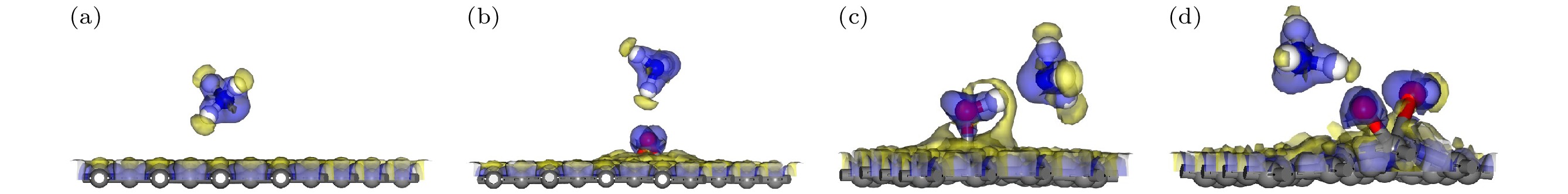
图 4 G和含有不同含氧基团的GO吸附NH3的差分电荷密度图(等值面为0.05) (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3
Figure 4. Charge density difference of G and GO with different oxygen-containing groups adsorbing NH3 (The isosurface value is 0.05): (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3.
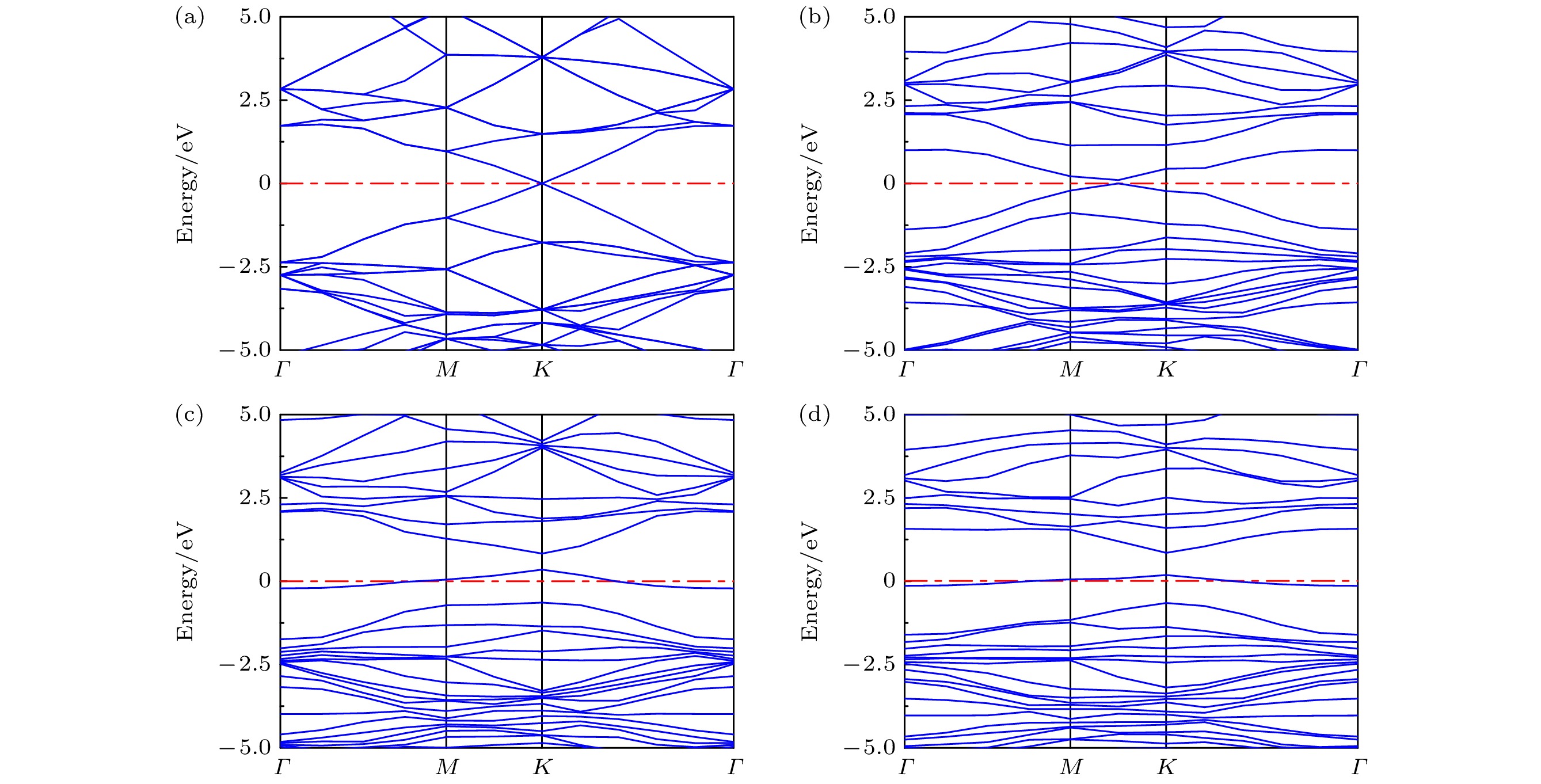
图 5 G和GO的能带结构 (a) G; (b) GO-O; (c) GO-OH; (d) GO-COOH
Figure 5. Band structures of G and GO: (a) G; (b) GO-O; (c) GO-OH; (d) GO-COOH.
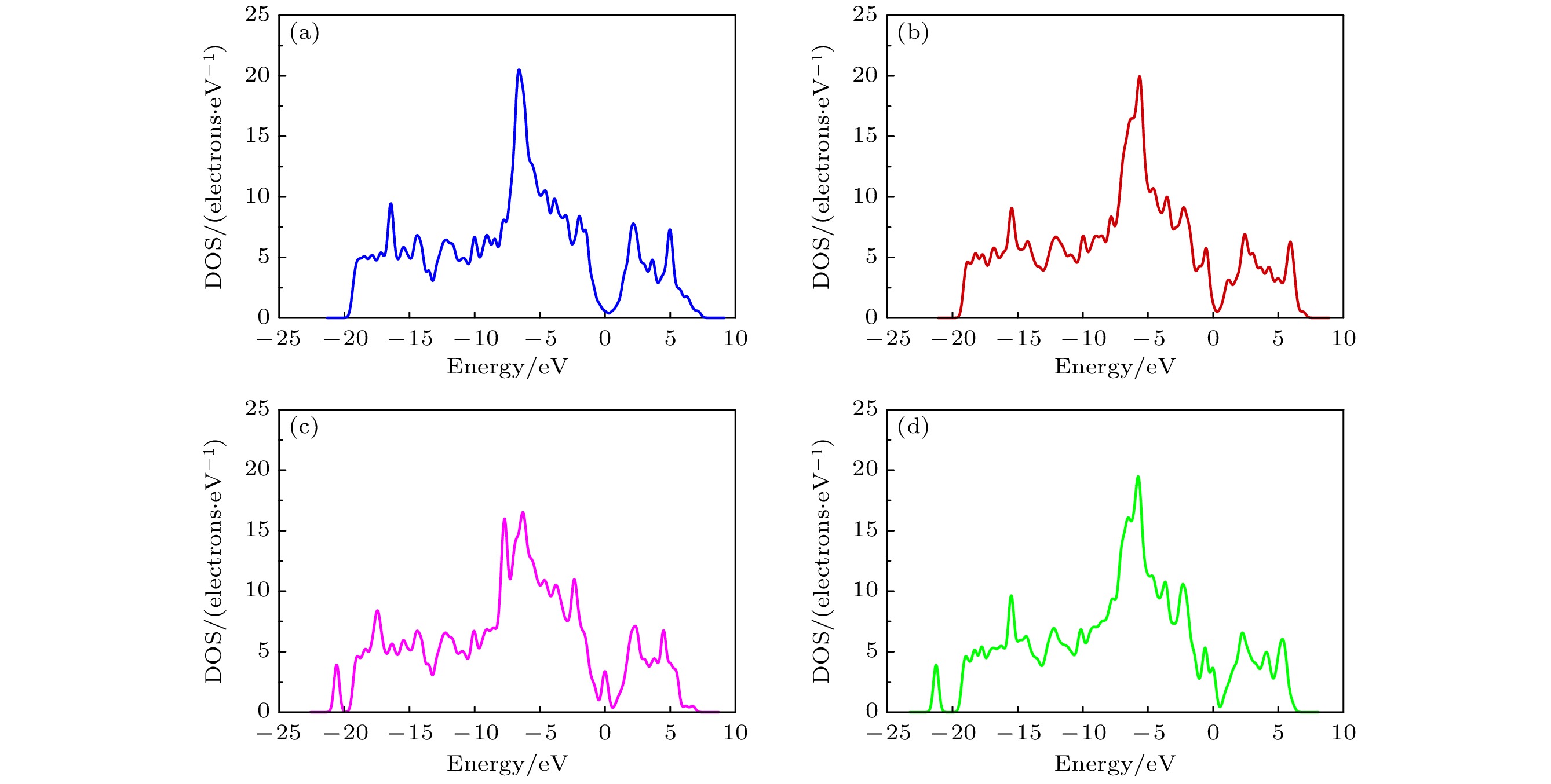
图 6 G和GO吸附NH3的态密度图 (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3
Figure 6. Density of states of G and GO adsorbing NH3: (a) G-NH3; (b) GO-O-NH3; (c) GO-OH-NH3; (d) GO-COOH-NH3.
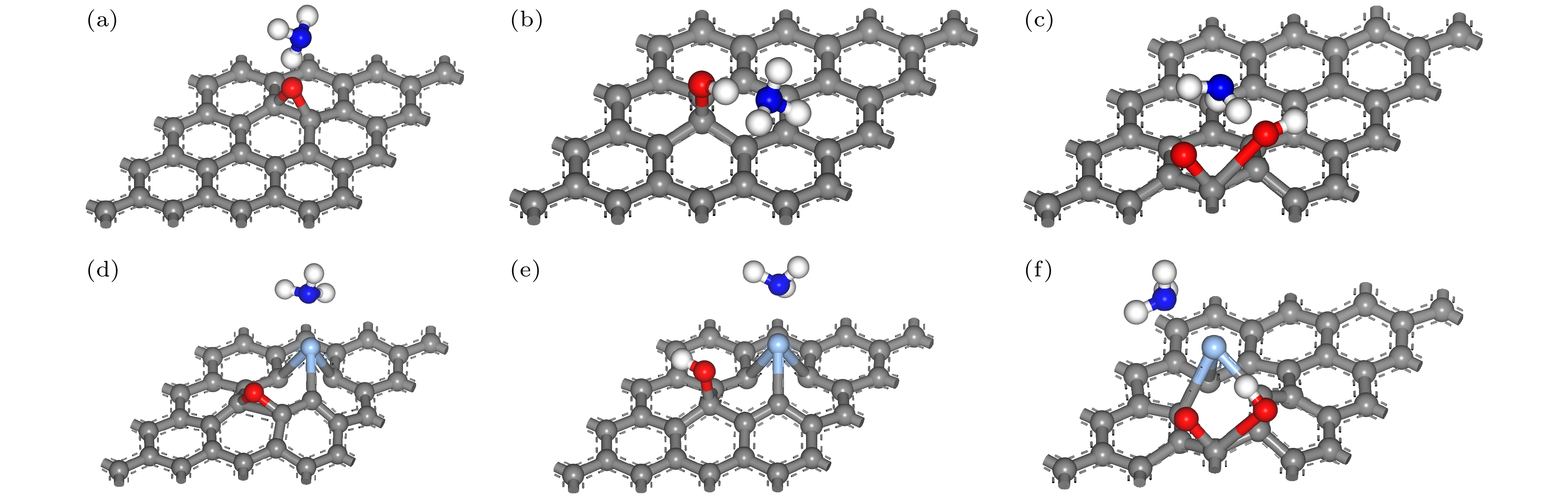
图 7 优化后含不同含氧基团的AgGO和GO吸附NH3的俯视图 (a) GO-O-NH3; (b) GO-OH-NH3; (c) GO-COOH-NH3; (d) AgGO-O-NH3; (e) AgGO-OH-NH3; (f) AgGO-COOH-NH3
Figure 7. Top views of optimized AgGO and GO with different oxygen-containing groups adsorbing NH3: (a) GO-O-NH3; (b) GO-OH-NH3; (c) GO-COOH-NH3; (d) AgGO-O-NH3; (e) AgGO-OH-NH3; (f) AgGO-COOH-NH3.
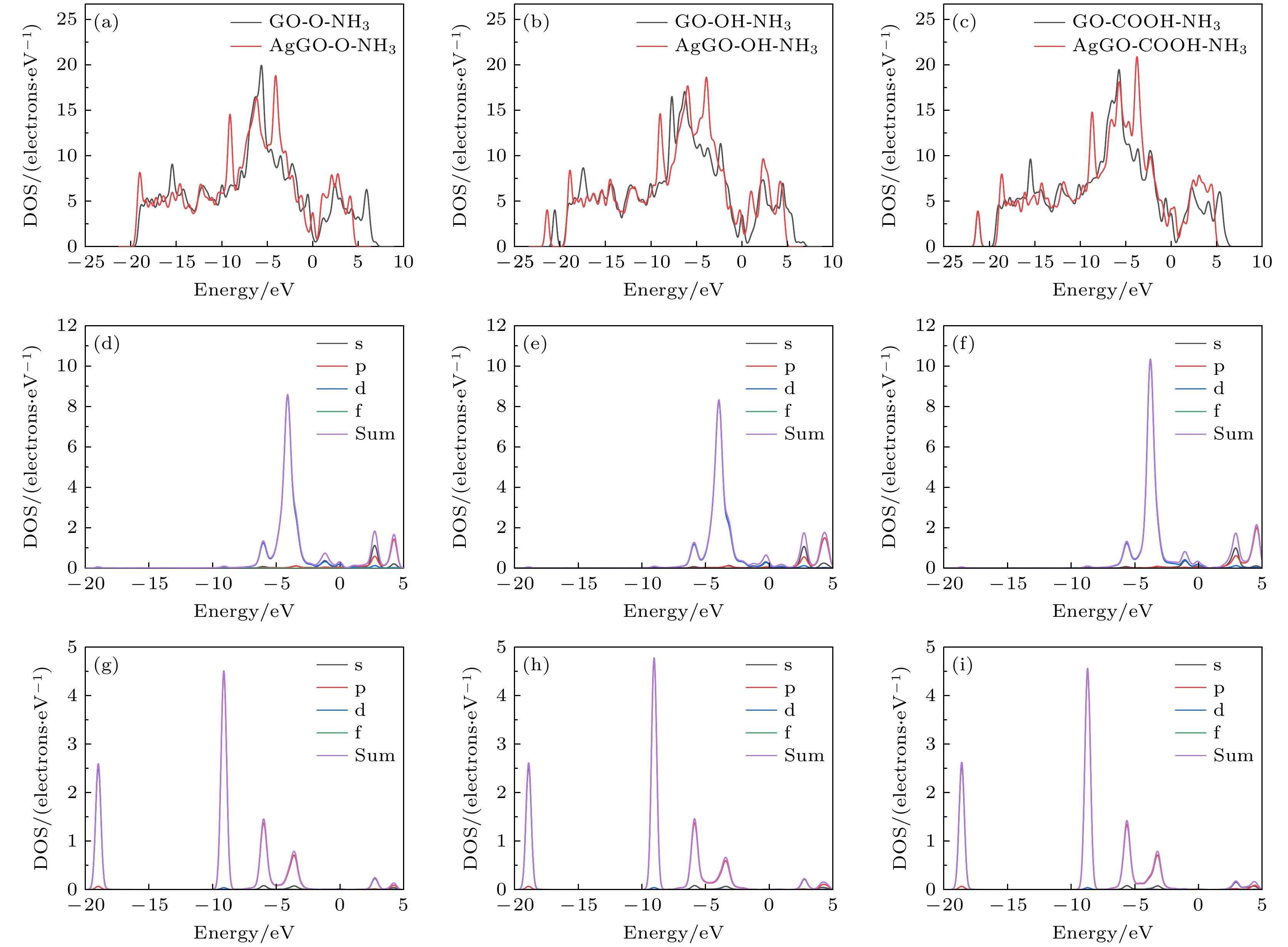
图 8 GO与AgGO的DOS和PDOS (a) GO-O-NH3和AgGO-O-NH3的DOS; (b) GO-OH-NH3和AgGO-OH-NH3的DOS; (c) GO-COOH-NH3和AgGO-COOH-NH3的DOS; (d) AgGO-O-NH3中Ag的DOS和PDOS; (e) AgGO-OH-NH3中Ag的DOS和PDOS; (f) AgGO-COOH-NH3中Ag的DOS和PDOS; (g) AgGO-O-NH3中N的DOS和PDOS; (h) AgGO-OH-NH3中N的DOS和PDOS; (i) AgGO-COOH-NH3中N的DOS和PDOS
Figure 8. DOS and PDOS of GO and AgGO: (a) DOS of GO-O-NH3 and AgGO-O-NH3; (b) DOS of GO-OH-NH3 and AgGO-OH-NH3; (c) DOS of GO-COOH-NH3 and AgGO-COOH-NH3; (d) DOS and PDOS of Ag in AgGO-O-NH3; (e) DOS and PDOS of Ag in AgGO-OH-NH3; (f) DOS and PDOS of Ag in AgGO-COOH-NH3; (g) DOS and PDOS of N in AgGO-O-NH3; (h) DOS and PDOS of N in AgGO-OH-NH3; (i) DOS and PDOS of N in AgGO-COOH-NH3.
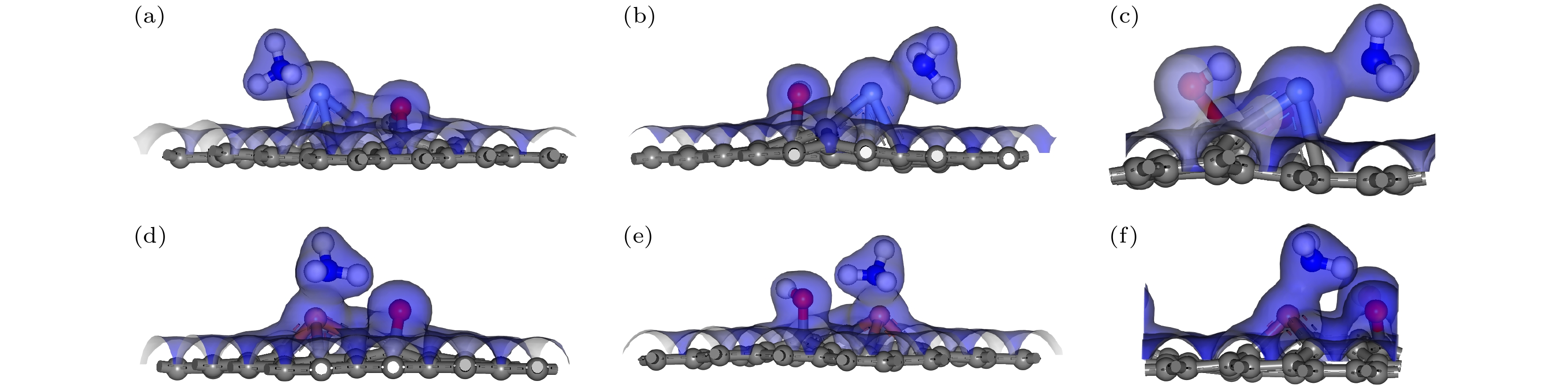
图 9 含有不同含氧基团的AgGO和CuGO吸附NH3的电荷总密度图(等值面为0.2) (a) AgGO-O-NH3; (b)AgGO-OH-NH3; (c) AgGO-COOH-NH3; (d) CuGO-O-NH3; (e) CuGO-OH-NH3; (f) CuGO-COOH-NH3.
Figure 9. Total charge density of AgGO and GO with different oxygen-containing groups adsorbing NH3 (The isosurface value is 0.2): (a) AgGO-O-NH3; (b) AgGO-OH-NH3; (c) AgGO-COOH-NH3; (d) CuGO-O-NH3; (e) CuGO-OH-NH3; (f) CuGO-COOH-NH3.
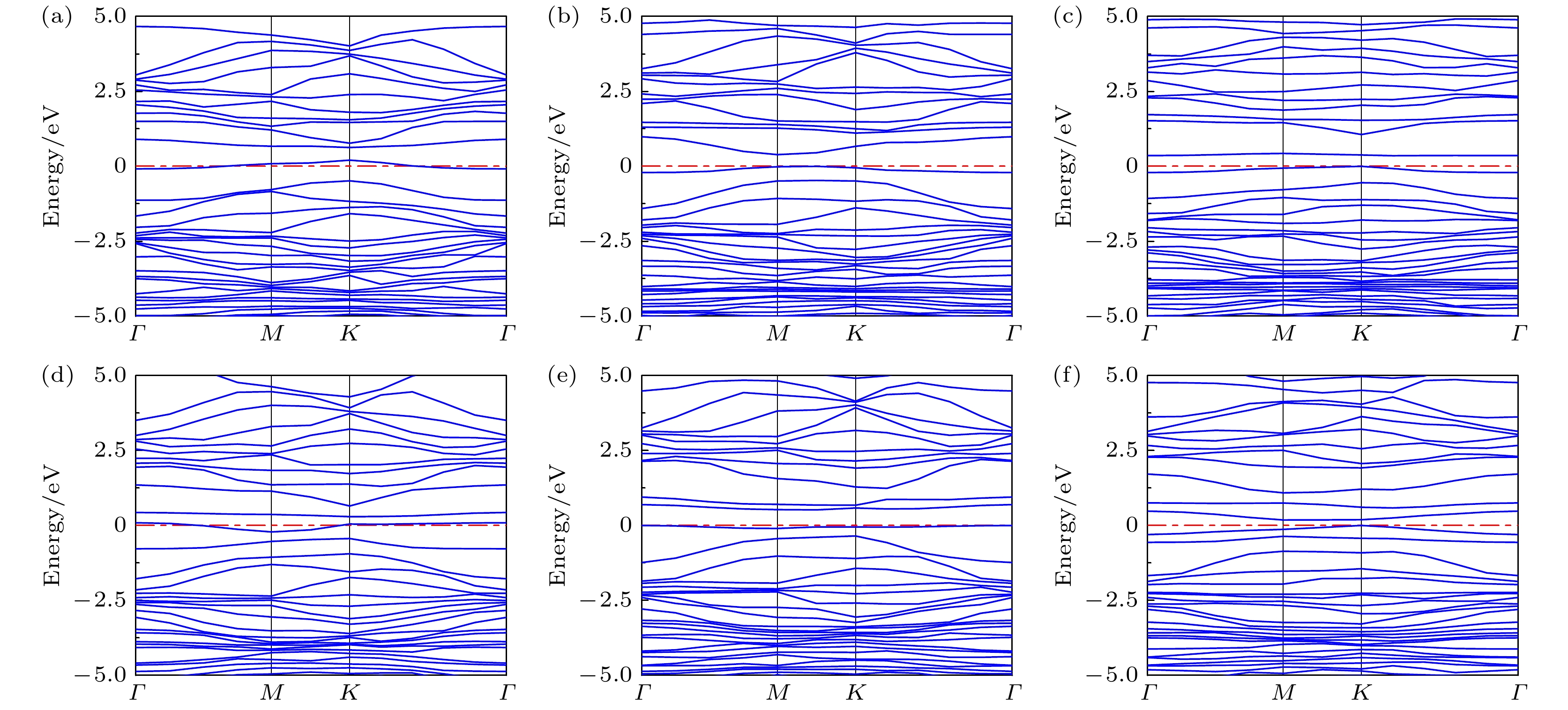
图 10 AgGO和CuGO的能带结构 (a) AgGO-O; (b) AgGO-OH; (c) AgGO-COOH; (d) CuGO-O; (e) CuGO-OH; (f) CuGO-COOH
Figure 10. Band structures of AgGO and CuGO: (a) AgGO-O; (b) AgGO-OH; (c) AgGO-COOH; (d) CuGO-O; (e) CuGO-OH; (f) CuGO-COOH.
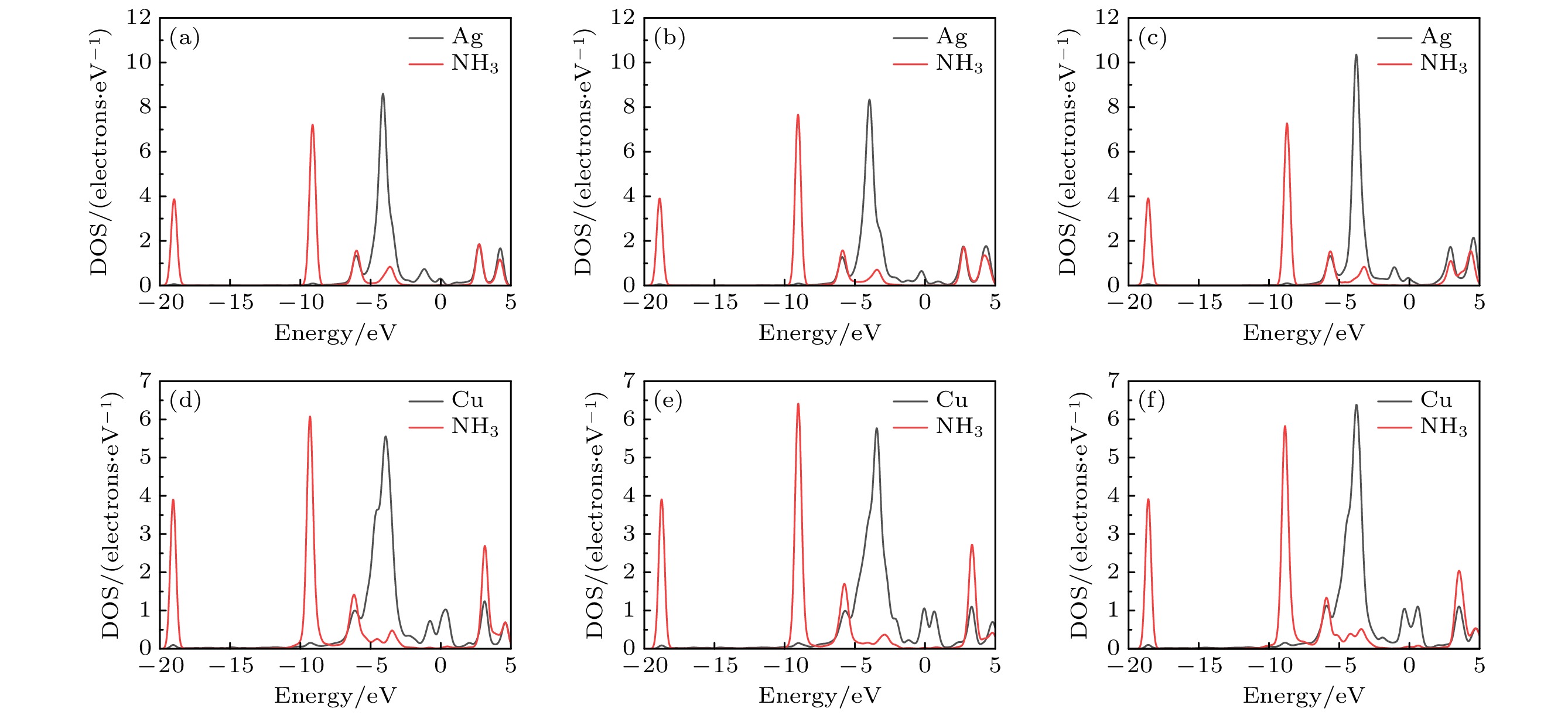
图 11 AgGO与CuGO的DOS (a) AgGO-O-NH3中Ag和NH3的DOS; (b) AgGO-OH-NH3中Ag和NH3的DOS; (c) AgGO-COOH-NH3中Ag和NH3的DOS; (d) CuGO-O-NH3中Cu和NH3的DOS; (e) CuGO-OH-NH3中Cu和NH3的DOS; (f) CuGO-COOH-NH3中Cu和NH3的DOS
Figure 11. DOS of AgGO and CuGO: (a) DOS of Ag and NH3 in AgGO-O-NH3; (b) DOS of Ag and NH3 in AgGO-OH-NH3; (c) DOS of Ag and NH3 in AgGO-COOH-NH3; (d) DOS of Cu and NH3 in CuGO-O-NH3; (e) DOS of Cu and NH3 in CuGO-OH-NH3; (f) DOS of Cu and NH3 in CuGO-COOH-NH3.
表 1 G和含有不同含氧基团的GO吸附NH3的几何参数. C1, C2和C3为与含氧基团相连的最近的三个碳原子; D表示吸附距离
Table 1. Structural parameters of G and GO with different oxygen-containing groups adsorbing NH3: C1, C2, and C3 are the three nearest carbon atoms connected to the oxygen-containing group; D represents the adsorption distance.
Species Bond angles/(°) D/Å C1-C2 C2-C3 C3-C1 G-NH3 120.06 119.98 119.94 3.24 (N—C) GO-O-NH3 118.75 118.19 118.17 2.28 (H—O) GO-OH-NH3 112.95 112.72 112.77 1.80 (N—H) GO-COOH-NH3 119.57 — — 2.44 (H—O)  下载: 导出CSV
下载: 导出CSV
表 2 G和含有不同含氧基团的GO吸附NH3的吸附能和电荷布居
Table 2. Adsorption energy and Mulliken charge of G and GO with different oxygen-containing groups adsorbing NH3.
System Mulliken charge/e Eads/eV C1 C2 C3 NH3 G-NH3 0.013 0.004 –0.039 0.022 –0.15 GO-O-NH3 0.112 0.024 0.026 0.005 –0.11 GO-OH-NH3 0.004 –0.035 0.006 0.078 –0.60 GO-COOH-NH3 0.125 –0.016 — 0.036 –0.17
下载: 导出CSV
表 3 带有不同含氧基团的AgGO和GO吸附NH3的吸附距离D、电荷转移Q和吸附能Eads
Table 3. Adsorption distance D, charge transfer Q, and adsorption energy Eads of AgGO and GO with different oxygen-containing groups adsorbing NH3.
Species D/Å Q/e Eads/eV NH3 Ag GO-O-NH3 2.28 (H-O) 0.005 — –0.11 AgGO-O-NH3 2.29 (N-Ag) 0.171 –0.038 –1.25 GO-OH-NH3 1.80 (N-H) 0.078 — –0.60 AgGO-OH-NH3 2.29 (N-Ag) 0.160 –0.033 –1.26 GO-COOH-NH3 2.44 (H-O) 0.036 — –0.17 AgGO-COOH-NH3 2.29 (N-Ag) 0.170 –0.082 –1.39
下载: 导出CSV
表 4 带有不同含氧基团AgGO和CuGO吸附NH3的电荷转移Q和吸附能Eads
Table 4. Charge transfer Q and adsorption energy Eads of AgGO and GO with different oxygen-containing groups adsorbing NH3.
Species Q/e Eads/eV NH3 Ag/Cu AgGO-O-NH3 0.171 –0.038 –1.25 CuGO-O-NH3 0.192 0.020 –1.39 AgGO-OH-NH3 0.160 –0.033 –1.26 CuGO-OH-NH3 0.180 –0.001 –1.43 AgGO-COOH-NH3 0.170 –0.082 –1.39 CuGO-COOH-NH3 0.192 0.034 –1.34
下载: 导出CSV
-
[1] Yu Z, Wang B, Li Y, et al. 2017 RSC Adv. 7 22599 doi: 10.1039/C7RA02740K [2] Hibbard T, Killard A J 2011 Crit. Rev. Anal. Chem. 41 21 doi: 10.1080/10408347.2011.521729 [3] Risby T H, Solga S F 2006 Appl. Phys. B 85 421 doi: 10.1007/s00340-006-2280-4 [4] Ishpal I, Kaur A 2013 J. Appl. Phys. 113 938 doi: 10.1063/1.4793994 [5] Wang J, Yang P, Wei X 2015 ACS Appl. Mater. Interfaces 7 3816 doi: 10.1021/am508807a [6] Li Y, Li H, Zhao F L 2024 Phys. Status Solidi RRL 18 2400015 doi: 10.1002/pssr.202400015 [7] Mirzaei M, Roohollahi H, Bagheri H 2024 Progresses in Ammonia: Science, Technology and Membranes (1st Ed.) (Amsterdam: Elsevier) pp69–94 [8] Kwak D, Lei Y, Maric R 2019 Talanta 204 713 doi: 10.1016/j.talanta.2019.06.034 [9] Zhu Y, Murali S, Cai W, Li X, Suk J W, Potts J R 2010 Adv. Mater. 22 3906 doi: 10.1002/adma.201001068 [10] Wu J, Lin H, Moss D J 2023 Nat. Rev. Chem. 7 162 doi: 10.1038/s41570-022-00458-7 [11] Bi J, Du Z, Sun J 2023 Adv. Mater. 35 2210734 doi: 10.1002/adma.202210734 [12] Schedin F, Geim A K, Morozov S V 2007 Nat. Mater. 6 652 doi: 10.1038/nmat1967 [13] Peng Y, Li J 2013 Front. Environ. Sci. Eng. 7 403 doi: 10.1007/s11783-013-0491-6 [14] Luo H, Zhang L, Xu S 2021 Appl. Surf. Sci. 537 147542 doi: 10.1016/j.apsusc.2020.147542 [15] Park M S, Kim K H, Kim M J 2016 Colloid Surface A 490 104 doi: 10.1016/j.colsurfa.2015.11.028 [16] Raza W, Krupanidhi S B 2018 ACS Appl. Mater. Interfaces 10 25285 doi: 10.1021/acsami.8b05162 [17] Tran Q T, Hoa H T M, Yoo D H 2014 Sens. Actuators, B 194 45 doi: 10.1016/j.snb.2013.12.062 [18] Karaduman I, Er E, Çelikkan H 2017 J. Alloys Compd. 722 569 doi: 10.1016/j.jallcom.2017.06.152 [19] Zhang L, Tan Q, Kou H 2019 Sci. Rep. 9 9942 doi: 10.1038/s41598-019-46213-9 [20] Saleh A M, Albiss B A 2024 ChemistrySelect 9 e202401500 doi: 10.1002/slct.202401500 [21] Li Q, Liu Y, Chen D 2021 Chemosensors 9 227 doi: 10.3390/chemosensors9080227 [22] Rawat S, Bamola P, Negi S 2023 ACS Appl. Nano Mater. 7 746 doi: 10.1021/acsanm.3c04787 [23] Sinnott S B 2013 J. Vac. Sci. Technol. , A 31 050812 doi: 10.1116/1.4813689 [24] Delley B 1990 J. Chem. Phys. 92 508 doi: 10.1063/1.458452 [25] Delley B 2000 J. Chem. Phys. 113 7756 doi: 10.1063/1.1316015 [26] Lerf A, He H, Forster M 1998 J. Phys. Chem. B 102 4477 doi: 10.1021/jp9731821 [27] Szabó T, Berkesi O, Forgó P 2006 Chem. Mater. 18 2740 doi: 10.1021/cm060258+ [28] Liu H, Liu Y, Zhu D 2011 J. Mater. Chem. 21 3335 doi: 10.1039/C0JM02922J [29] Guo B, Fang L, Zhang B 2011 Insciences J. 1 80 doi: 10.5640/insc.010280 [30] Geim A K, Novoselov K S 2007 Nat. Mater. 6 183 doi: 10.1038/nmat1849 [31] Wei D, Zhao C, Khan A 2019 Chem. Eng. J. 375 121964 doi: 10.1016/j.cej.2019.121964 [32] Yan J A, Chou M Y 2010 Phys. Rev. B 82 125403 doi: 10.1103/PhysRevB.82.125403 [33] 王晓, 黄生祥, 罗衡, 邓联文, 吴昊, 徐运超, 贺军, 贺龙辉 2019 物理学报 68 187301 doi: 10.7498/aps.68.20190523 Wang X, Huang S X, Luo H, Deng L W, Wu H, Xu Y C, He J, He L H 2019 Acta. Phys. Sin. 68 187301 doi: 10.7498/aps.68.20190523 [34] Giovannetti G, Khomyakov P A, Brocks G 2008 Phys. Rev. Lett. 101 026803 doi: 10.1103/PhysRevLett.101.026803 -
计量
- 文章访问数: 195
- HTML全文浏览数: 195
- PDF下载数: 7
- 施引文献: 0