
 首页
首页 登录
登录 注册
注册



-
人类社会不可阻挡地向信息化、智能化方向发展, 对能源存储和能量转化提出了更高的要求. 太阳能、风能、潮汐能、地热能等可再生能源的间歇性和随机性促进了储能系统的发展[1–3]. 电化学储能因其高能量密度、高循环效率、应用灵活等优点被认为是一种理想的储能方式. 在涉及电子设备和电机的许多应用中[4,5], 可充电电池作为主要的储能技术, 凭借其较高的能量密度、优异的能源效率和较长的循环寿命, 已成为替代传统能源存储技术的主要方向之一[6–9], 尤其是钠离子电池(sodium-ion batteries, SIBs)和锂离子电池(lithium-ion batteries, LIBs), 在电能存储领域的应用潜力日益突出[10]. 可充电电池的性能在很大程度上取决于电极材料, 特别是负极材料的组成、结构和性能. 因此, 开发高性能的负极材料已成为推进电池技术发展的关键方向之一. 在众多的电极材料中, 二维材料因其优异的比表面积、卓越的机械强度和可调节的化学活性, 逐渐受到储能和电催化领域研究人员的广泛关注[11–14], 其中MBene材料备受瞩目. 自2017年Guo等[15]通过理论预测提出Mo2B2和Fe2B2这两种二维MBene, 并评估其作为LIBs电极材料的潜力以来, 越来越多的MBene材料通过理论计算被设计和探索. Mo2B凭借其良好的导电性、导热性、较大的存储容量和低扩散势垒, 在LIBs中展现出了令人满意的电化学性能[16]. 同时, Sc2B, Ti2B和V2B也被预测为镁离子电池(magnesium-ion batteries, MIBs)电极材料的潜在候选者[17]. V2B2, Cr2B2和Mn2B2被认为是LIBs和SIBs电极材料的潜在候选者[18].
此外, MBenes表面官能团的选择也是实现高性能电极材料的关键因素之一. 一般来说, MBenes表面裸露的金属原子由于存在大量悬挂键, 具有较高的化学活性, 因此在化学合成过程中, 容易在其表面引入官能团[19]. Khaledialidusti等[20]发现与原始六方MBene相比, O和OH官能团化的六方MBene具有更优异的力学性能. Liang等[21]报告了N官能团化的Ti2B, 研究表明 Ti2BN2是SIBs负极材料的潜在候选者. 正如前述, MBenes的选择性官能团化对高性能电极材料的开发至关重要. 其中, S也被认为是一种潜在的表面终端, 可以有效调节金属离子的吸附能力, 从而实现较高的理论比容量[22,23]. 据Wang等[24]报道, S官能团化的Ti2B表现出良好的结构稳定性和优异的SIBs电化学性能.
鉴于MBenes表面官能团化对电极材料性能的显著影响, 本研究重点探讨了基于Zr2B和Nb2B基底的两种全新MBene材料的表面官能团化、结构稳定性、电子性质、金属离子吸附性能及其扩散势垒、理论比容量、开路电压等电化学性能, 特别是官能团化结构在LIBs和SIBs中的应用潜力. 研究结果表明, Zr2BS2和Nb2BS2材料不仅具有优异的稳定性, 还表现出较高的金属离子吸附能力和较低的扩散势垒, 具有显著的理论比容量和倍率性能, 预示着它们在SIBs中广阔的应用前景. 通过本研究, 我们旨在为开发新型高效、稳定的M2B型MBene LIBs和SIBs负极材料提供理论依据, 并为未来高性能LIBs和SIBs的设计与应用提供指导.
-
本文所有计算均采用密度泛函理论框架下的VASP(Vienna ab-initio simulation package)[25,26]进行. 采用投影缀加波(projected augmented wave)方法描述原子核、内层电子与自由电子之间的相互作用[27,28]. 选择Perdew-Burke-Ernzerhof泛函中的广义梯度近似来描述交换和关联函数[29,30]. 在所有计算中, 均采用色散矫正策略(DFT-D3)描述范德瓦耳斯相互作用, 以考虑非键合力的影响[31]. 晶胞结构优化基于Gamma中心9×9×1, 12×12×1和3×3×1的Monkhorst-Pack网格分别对Zr2B, Nb2B原胞和3×3×1超胞的布里渊区进行采样, 原子力和能量收敛标准为10–5 eV/Å 和10–7 eV, 平面波截断能设定为500 eV. 为避免周期性边界条件引起的相互作用, 沿垂直平面方向设置了厚度为20 Å的真空层. 采用PHONOPY程序包中实现的密度泛函微扰理论(DFPT)方法计算了声子色散曲线, 以研究MBene的动力学稳定性[32,33]. 此外, 在350 K下使用正则(NVT)系综进行了从头算分子动力学模拟(ab-initio molecular dynamics, AIMD), 每次模拟持续10 ps, 步长为1 fs, 以研究结构的热力学稳定性. 并采用CI-NEB(climbing image nudged elastic band)方法, 研究金属原子在基底上的扩散行为并计算扩散势垒[34], 其中初末态之间插入3个中间点以构建扩散路径, 相邻图像间原子的平均位移距离为0.87 Å.
-
基底M2B(M = Zr, Nb)单层属于
$ P\bar 3 m1 $ 空间群, 呈现出典型的M-B-M层状三明治结构, 如图1所示. 为了探索基底M2B单层表面官能团化原子的位置依赖性, 本研究基于能量最低化原则, 系统研究了3种不同的官能团化位点, 如图1(a), (b)所示, 即位点I (B顶位)、位点II (M1顶位)和位点III (M2顶位). S原子的最终优化配置如图1(c), (d)所示, 其倾向于占据位点III, 与其他基于MXene的S官能团化材料(Ti2CS2[35], V2CS2[36])中的S原子分布规律相一致. 这一现象表明, 位点III是M2B单层表面官能团化的优选位置.材料的结构稳定性是电池应用中的基本要求. 为了评估材料的动力学和热力学稳定性, 本研究采用了声子谱分析和AIMD方法. 首先, 对基底结构进行了稳定性评估. 由图2(a), (b)所示, Zr2B和Nb2B基底在整个布里渊区内均未出现虚频, 表明两种基底具有良好的动力学稳定性. 此外, 采用NVT系综在350 K下进行了时长为10 ps、步长为1 fs的AIMD模拟, 验证其热力学稳定性, 结果如图2(c), (d)所示. 随后, 为了确认S官能团化对材料稳定性的影响, 针对S官能团化结构进行了AIMD模拟计算, 确保其在电池运行环境下的稳定性, 结果如图2(e), (f)所示. 模拟结果表明, 所有结构均未出现化学键断裂或重连现象, 并且自由能沿中心轴呈现稳定的振荡, 表明其在热力学上具有良好的稳定性. 这表明该材料在相对高温下具有较强的工作潜力, 能够满足电池应用中的稳定性要求.
-
可充电金属离子电池的循环性能、倍率性能和电导率等关键参数与电极材料的电子性质密切相关. 因此, 为了系统评估两种材料的电子性质, 本研究通过计算能带结构和态密度进行分析. 如图3(a), (b)所示, 费米能级(0 eV)穿过Zr2BS2和Nb2BS2的能带, 表明这两种材料具有优异的金属性. 此外, 自旋向上和自旋向下的能带完全重叠, 表明两者均表现出非磁性特征. 态密度分析结果如图3(c), (d)所示, 费米能级附近存在显著的态密度峰, 表明两种材料具有较高的载流子密度, 这对于电荷转移过程中的载流子补充至关重要. 分波态密度进一步表明, 金属性能主要来源于过渡金属元素Zr和Nb, 这一特征与其他典型负极材料类似, 如Cr2CS2, Mo2CS2[37]等. 综上所述, 这两种官能团化材料的优异金属性质确保了其出色的导电性和较高的载流子密度. 金属性的优势有助于减少电池工作过程中产生的欧姆热和降低导电添加剂的需求, 此外, 较高的载流子密度通常意味着更高的电子导电性, 有助于支持快速充放电并减小能量损失. 以上特性表明, 这些材料在作为金属离子电池电极材料方面具有显著的应用潜力.
-
金属离子的吸附或嵌入可能引起电极材料晶格体积的膨胀. 在充放电过程中, 电极材料会经历反复的体积变化, 如果体积变化过大, 可能导致材料的塌陷或粉化, 从而影响电池的循环稳定性. 因此, 电极材料需要具备良好的力学性能, 能够在应力作用下有效承受应变而不发生断裂. 为评估Zr2BS2和Nb2BS2的力学性能, 通过VASP计算了材料的弹性常数(C )、杨氏模量(E )和泊松比(ν), 并且通过不断增大K点密度的方法来使弹性常数计算收敛(波动不超过5 N/m), 其中, MoS2的计算结果与Singh等[38]的计算一致, 一定程度上验证了计算参数设置的合理性, 结果如表1所列. 杨氏模量和泊松比通过(1)式、(2)式计算[39]:
其中Cₓₓ, Cyy, Cₓy分别表示沿x, y和xy方向的弹性常数, 根据Born-Huang准则[40], 稳定的二维材料需要满足: C11C12 > 0,
$ {C_{11}}{C_{22}} - C_{12}^2 > 0 $ , C66 > 0, 由表1可知, Zr2BS2和Nb2BS2均具备力学稳定性. 并且与MoS2相比, 均具有较高的杨氏模量和适中的泊松比, 这表明, 两种材料在力学性质方面表现出良好的刚性和弹性, 具有较强的抗应变能力, 因此在充放电过程中能够承受较大的体积变化而不发生结构破坏, 具备良好的力学稳定性, 适合用作金属离子电池电极材料. -
金属离子在材料表面的优异吸附性能是电池稳定运行的关键因素之一. 因此, 本研究探讨了单个金属离子(Li+, Na+)在官能团化材料表面的吸附行为. 首先, 我们构建了3×3×1的M2BS2(M = Zr, Nb)超晶胞, 提出了两个潜在的吸附位点, 分别为位点α(B顶位)、位点β(M顶位), 并计算了各位点的吸附能, 如图4(a), (b)所示. 基于能量最低化原则, 我们发现对于Zr2BS2, Li+和Na+的最佳吸附位点为α位点, 而对于Nb2BS2, 则是β位点. 为了深入理解金属离子的吸附特性, 进一步分析了金属离子吸附模型的差分电荷密度Δρ (
$ \Delta \rho = {\rho _{{\text{system}}}} - {\rho _0} $ , 其中ρsystem为吸附后的体系电荷密度, ρ0为未吸附时基底材料的电荷密度), 结果如图4所示, 其中暖色(红/黄)区域表示电荷积累, 冷色(蓝/绿)区域表示电荷耗散. 对比分析表明, 在Zr2BS2体系中, Li+/Na+在α位点的吸附诱导出显著高于β位点的电荷密度积累($ \Delta {\rho _\alpha } > {\text{ }}\Delta {\rho _\beta } $ ), 而在Nb2BS2体系中则呈现相反趋势($ \Delta {\rho _\beta } > {\text{ }}\Delta {\rho _\alpha } $ ), 这种位点依赖的电荷再分布特征从电子结构层面揭示了不同材料对Li+/Na+吸附位点选择性的本质差异, 并且表明金属离子通过与官能团原子之间的相互作用形成了化学键. 这种相互作用赋予了金属离子在官能团化材料表面良好的吸附性能. 进一步, 为了探究Li+/Na+在Zr2BS2和Nb2BS2负极的反应形式, 在350 K下进行了8 ps的AIMD模拟, 结果如图5所示. 可以观察到, 在8 ps内所有结构均没有明显的结构破坏和LixSy的形成, 证明Li+/Na+的负极反应形式确为吸附式, 而非合金化、插入式或者2H-MoS2[41]般的转换式. 综上所述, 金属离子与材料表面之间的电荷分布变化揭示了静电相互作用在吸附过程中的重要作用. 金属离子通过静电吸引力与表面官能团相结合, 从而实现了有效的吸附, 确保了电极材料在充放电过程中的稳定性和高效性能. -
电极材料表面吸附原子的扩散速率是影响电池充放电速率的关键因素之一. 因此, 本研究系统地探讨了Li+, Na+在Zr2BS2和Nb2BS2表面的扩散特性. 每种金属离子在两种材料表面都具有3条可能的扩散路径. 如图6(a), (b), 对于Zr2BS2, 扩散路径分别为: 路径I (a→a')、路径II (a→b→a')和路径III (a→c→a'); 对于Nb2BS2, 扩散路径分别为: 路径I (x→x')、路径II (x→y→x')和路径III (x→z→x'). 然而, 在计算过程中发现, 对于两种材料, 路径I会自动转化为路径II, 因此我们仅对路径II和路径III进行了进一步的讨论. 从图6(c)—(f)可以看出, 路径III的能垒显著高于路径II, 这表明路径II为Zr2BS2和Nb2BS2表面金属离子的最佳扩散路径. 具体而言, 对于Zr2BS2, Li+和Na+的扩散势垒分别为0.233 eV和0.131 eV, 而对于Nb2BS2, Li+和Na+的扩散势垒分别为0.211 eV和0.088 eV. 可以看出, Na+的扩散势垒均低于Li+, 这意味着Na+在Zr2BS2和Nb2BS2表面具有更高的迁移速率, 符合与其他MXene相关材料(例如MoN2[42])观察到的相似趋势. 更为重要的是, Zr2BS2和Nb2BS2的扩散势垒相较于其他二维材料, 如Mn2B2[18] (Li+/Na+为0.29 eV/0.17 eV)和MoN2[42] (Li+/Na+为0.78 eV/0.56 eV), 具有显著的优势, 表明这两种材料在电池应用中具有优异的倍率性能, 预示着它们能在较高的充放电速率下工作, 还表明Zr2BS2和Nb2BS2作为电池电极材料具有巨大的应用潜力.
-
金属离子存储容量是评估高性能可充电金属离子电池电极材料的重要指标. 在电池充放电过程中, 金属离子不断从正极脱嵌并嵌入负极, 负极材料对金属离子的存储容量直接决定了电池的整体容量. 大量的理论计算表明[43–46], 在许多MXene和MBene类材料的表面, 金属离子能够进行多层吸附, 从而显著提升材料的比容量. 受到这一现象的启发, 本研究探讨了Li+, Na+在Zr2BS2和Nb2BS2表面逐层吸附的可能性, 并考虑了不同层之间的最佳吸附位点. 计算结果表明, Zr2BS2和Nb2BS2在吸附第2层Li+与第4层Na+时平均吸附能转变为正值(对于Zr2BS2, Li+/ Na+分别为0.296 eV/0.020 eV, 对于Nb2BS2, Li+/ Na+分别为0.050 eV/0.054 eV), 即Zr2BS2和Nb2BS2均只能够在其表面吸附一层Li+和三层Na+. 为了进一步理解金属离子与官能团化MBene表面之间的结合特征及其多层吸附行为, 进行电子局域化函数(electron localization function, ELF)计算. ELF值表示在参考电子附近找到具有相同自旋电子的概率, 取值范围为0—1, 其中ELF = 1表示电子完全局域化, ELF = 0.5表示电子气型的成对概率, ELF = 0表示电子完全离域化. 在讨论金属离子的逐层吸附时, 负电子云(negative electron cloud, NEC)的形成是吸附稳定性的关键因素, 其有助于进一步吸引外层离子. 因此, 我们绘制了吸附第一层Li+, Na+时(1 1 0)截面的ELF图. 如图7所示, 电子在第1层Li+, Na+与Zr2BS2, Nb2BS2基底之间局域化, 验证了金属离子在基底上的稳定吸附. 此外, 在Na+层上方形成了完整的NEC层, 不仅确保了第1层Na+的稳定吸附, 还增强了对额外Na+的吸引力, 进一步证实了Zr2BS2和Nb2BS2能够实现Na+的多层吸附, 这与我们的计算结果一致.
基于金属离子的吸附能力, 使用以下公式计算了金属离子电池的理论比容量(Cm)[47]:
其中, Xmax表示吸附金属离子的最大浓度, 对于单层和三层吸附, Xmax分别为2和6; i表示吸附阳离子的价数; F表示法拉第常数(26801 mA·h/mol);
$ {m_{{M_{2}}{\text{B}}{{\text{S}}_{2}}}} $ 为M2BS2(M = Zr, Nb)的摩尔质量. 计算结果显示, 对于LIBs, Zr2BS2和Nb2BS2的理论比容量分别为208 mA·h/g和205 mA·h/g, 而Zr2BS2和Nb2BS2在SIBs中的理论比容量分别为624 mA·h/g和616 mA·h/g, 显著高于其他二维电极材料, 如Ti2B2(342 mA·h/g), Cr2B2(491.8 mA·h/g)和 Mn2B2(482.9 mA·h/g)等[18]. 综上所述, 尽管Zr2BS2和Nb2BS2不适合用作LIBs电极材料, 但由于它们对Na+具有较高的存储容量, 这使得它们有可能成为性能优异的SIBs电极材料. -
开路电压(open circuit voltage, OCV)是评估新型离子电池负极材料潜在适用性的关键参数之一. 鉴于Zr2BS2和Nb2BS2在LIBs中的理论比容量较低, 不适用于LIBs应用. 因此, 本研究仅考虑了Zr2BS2和Nb2BS2在SIBs中的OCV值. M2BS2(M = Zr, Nb)的充放电过程可描述为: M2BS2 + x·e– + x ·Na+ ↔ M2BS2Naₓ. 通常, 通过计算Na+吸附前后的总能量差异来确定OCV值. 我们忽略了体积变化项(PΔV )和熵项(TΔS ), 因此, 内能的变化近似等于吉布斯自由能的变化[48]. 根据这一近似, OCV值可用(4)式表示:
其中,
$ {E_{{M_{2}}{\text{B}}{{\text{S}}_{2}}}} $ , ENa 和$ {E_{{M_{2}}{\text{B}}{{\text{S}}_{2}}{+}n{\text{Na}}}} $ 分别表示M2BS2、体相中单个Na原子的总能量以及M2BS2吸附n个Na+后系统的总能量; n表示吸附Na+的数量(对于单层吸附, n = 18; 对于三层吸附, n = 54); x表示电解质中Na+所带的电荷. 计算结果如图8所示, Na+在Zr2BS2和Nb2BS2表面吸附的OCV值分别为0.3808 V和0.2102 V. 通常, 较低的OCV值(小于1 V)有助于减少枝晶的形成, 从而提高电池的安全性[49,50]. 综上所述, Zr2BS2和Nb2BS2在SIBs中的OCV值表明它们具有较好的安全性和稳定性, 适合作为SIBs负极材料. -
本文设计构建了基于Zr2B和Nb2B基底的S官能团化MBene材料Zr2BS2和Nb2BS2, 并通过声子色散曲线、AIMD模拟、弹性常数证明其动力学、热力学以及力学稳定性. 倍率性能方面, 首先通过能带结构与态密度显示了Zr2BS2和Nb2BS2的优异导电性, 其次两种材料对于SIBs分别具有0.131 eV和0.088 eV的低扩散势垒, 表明它们在高倍率充放电过程中的高效性. 能量存储能力方面, Zr2BS2和Nb2BS2在SIBs中的理论比容量分别达到624 mA·h/g和616 mA·h/g, 远超许多其他二维电极材料, 展示了它们在SIBs领域的广阔应用前景. 安全性能方面, 金属离子的吸附性能分析表明, Zr2BS2和Nb2BS2对Na+具有较强的吸附能力, 进一步增强了它们在电池充放电过程中的循环稳定性. 此外, Zr2BS2和Nb2BS2的OCV值分别为0.3808 V和0.2102 V, 较低的OCV值有助于减少枝晶的形成, 提升电池的安全性. 综合来看, 本研究不仅为MBene基负极材料的设计提供了理论框架, 还通过揭示表面官能团化、结构稳定性与离子传输的关联, 为其他二维材料及非二维体系的设计提供了重要借鉴, 并且有助于理解其他TMD基LIBs/SIBs的充放电机理. 未来需进一步探索实验合成路径、规模化制备技术及全电池性能验证, 推动此类材料从理论向实际应用的跨越.
MBene基高性能离子电池负极材料的第一性原理研究
First-principles study of MBene-based high-performance anode materials for ion batteries
-
摘要: 二维过渡金属硼化物(MBene)作为新型金属离子电池电极材料, 具有MB, M2B, M2B2等多种相结构, 然而现有研究对于M2B相体系的探索仍显匮乏. 本研究聚焦于M2B相MBene的设计, 首次构建了硫(S)官能团化的Zr2BS2和Nb2BS2两种全新材料, 系统揭示了其作为锂/钠离子电池负极材料的性能机制. 通过第一性原理的计算方法, 证实Zr2BS2和Nb2BS2两种材料具备优异的结构稳定性, 并且在钠离子电池中展现出较高的理论比容量(分别为624 mA·h/g和616 mA·h/g)以及较低的扩散势垒(Na+扩散势垒低至0.131 eV和0.088 eV). 同时, 其较低的开路电压(0.38 V和0.21 V)可有效抑制枝晶生长, 兼具高容量与安全性. 本研究不仅完善了M2B相MBene体系的系统性研究, 更为开发高容量、快充型钠离子电池负极材料提供了理论指导.Abstract: Two-dimensional transition metal borides (MBene), as emerging electrode materials for metal-ion batteries, exhibit various phase structures, including MB, M2B, and M2B2. However, current research on the M2B-phase system remains insufficient. This study focuses on the design of M2B-phase MBenes, pioneering the construction of two novel sulfur-functionalized materials, Zr2BS2 and Nb2BS2, while systematically elucidating their performance mechanisms as anode materials for lithium/sodium-ion batteries. Through first-principles calculations, both Zr2BS2 and Nb2BS2 demonstrate exceptional structural stability and superior electrochemical properties in sodium-ion battery applications. Specifically, they exhibit high theoretical specific capacities (624 mA·h/g and 616 mA·h/g) and remarkably low diffusion energy barriers for Na+ (0.131 eV and 0.088 eV). Moreover, their low open-circuit voltages (0.38 V and 0.21 V) effectively suppress dendrite growth, achieving an optimal balance between high capacity and operational safety. This work not only establishes a theoretical framework for MBene-based anode design but also provides critical insights into the correlation between surface functionalization, structural stability, and ion transport kinetics. These findings provide valuable guidance for developing other two-dimensional materials and non-layered systems, while contributing to mechanistic understanding of charge-discharge processes in transition metal dichalcogenide TMD-based lithium/sodium-ion batteries.
-

-
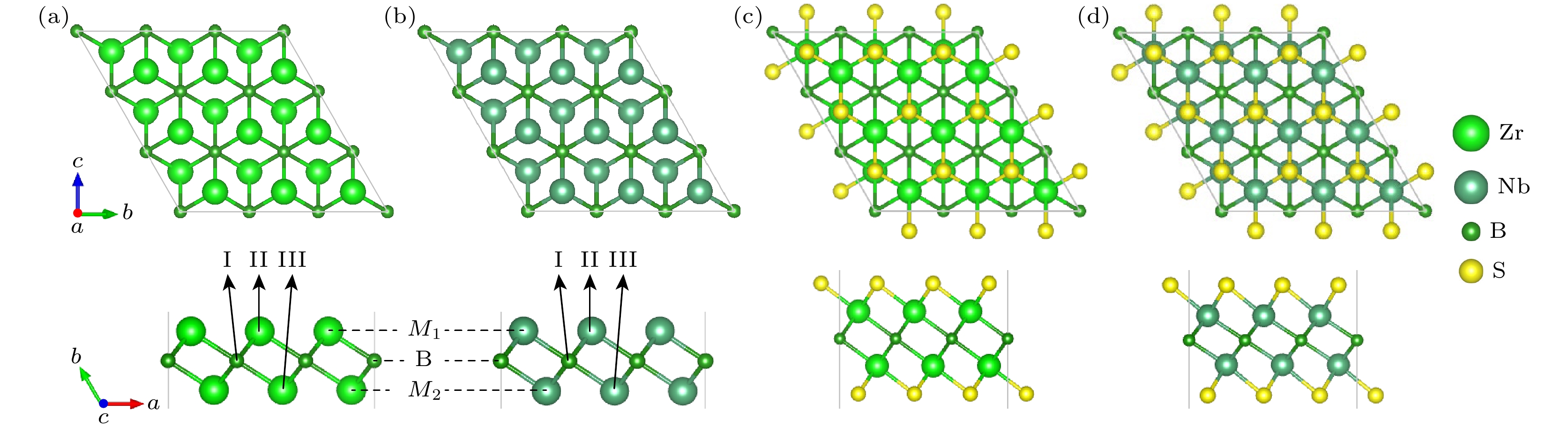
图 1 (a) Zr2B, (b) Nb2B, (c) Zr2BS2和(d) Nb2BS2 结构模型
Figure 1. Structural models of (a) Zr2B, (b) Nb2B, (c) Zr2BS2, and (d) Nb2BS2.
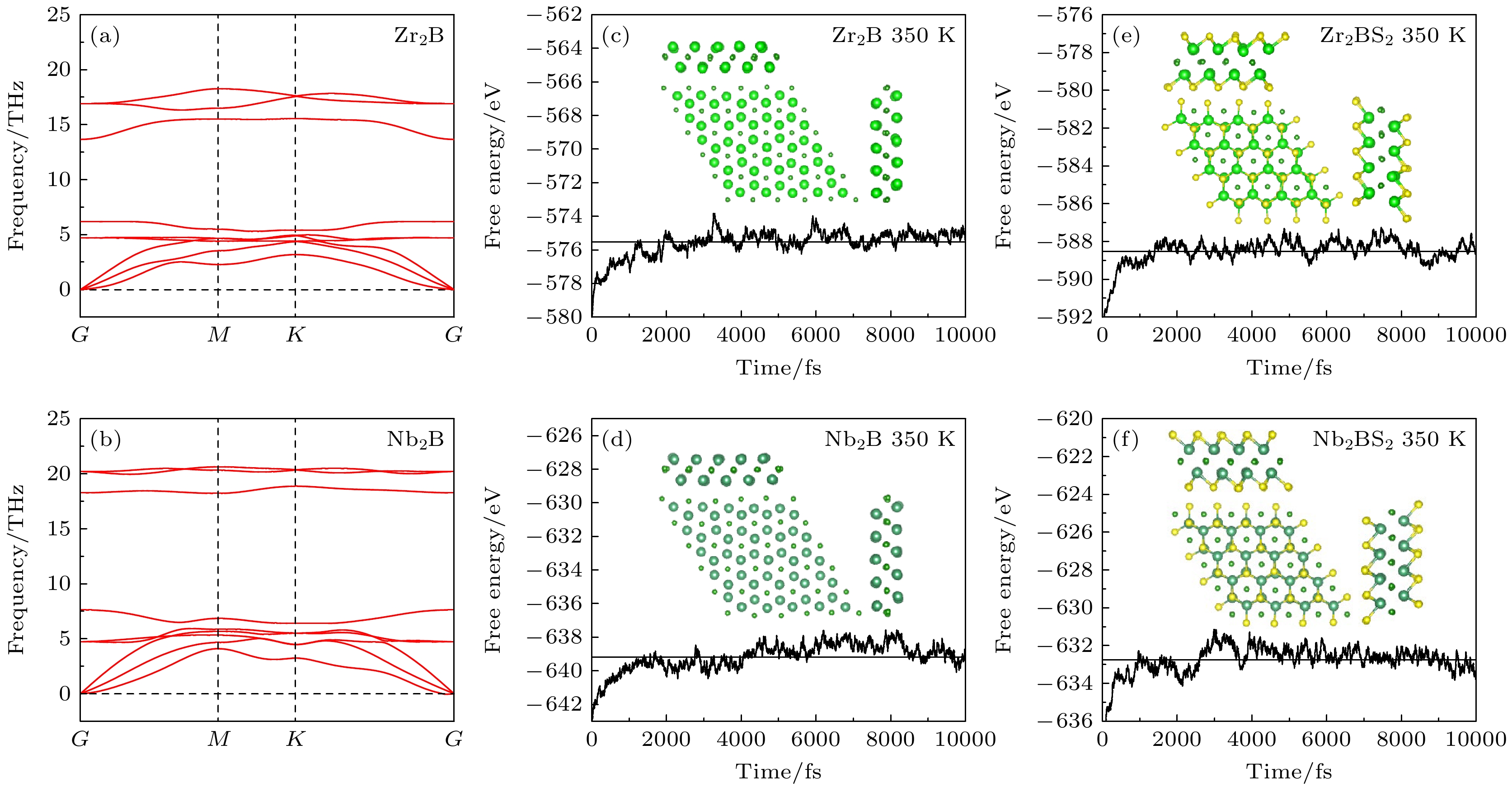
图 2 (a) Zr2B和(b) Nb2B声子色散曲线; 350 K下(c) Zr2B, (d) Nb2B, (e) Zr2BS2和(f) Nb2BS2的AIMD模拟
Figure 2. Phonon dispersion curves of (a) Zr2B and (b) Nb2B; AIMD simulations at 350 K for (c) Zr2B, (d) Nb2B, (e) Zr2BS2, and (f) Nb2BS2.
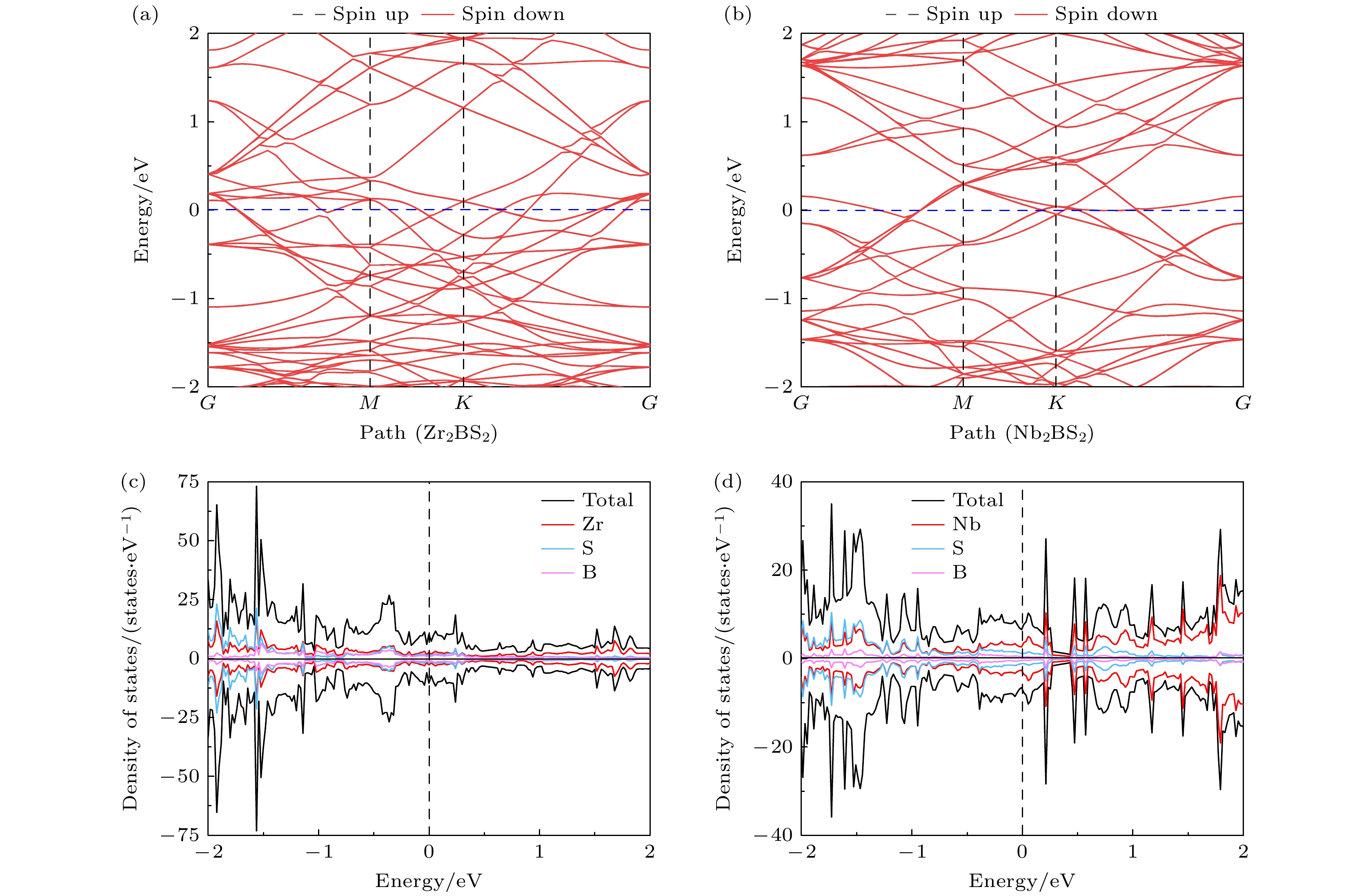
图 3 (a) Zr2BS2和(b) Nb2BS2能带结构; (c) Zr2BS2和(d) Nb2BS2态密度
Figure 3. Band structures of (a) Zr2BS2 and (b) Nb2BS2; DOS of (c) Zr2BS2 and (d) Nb2BS2.
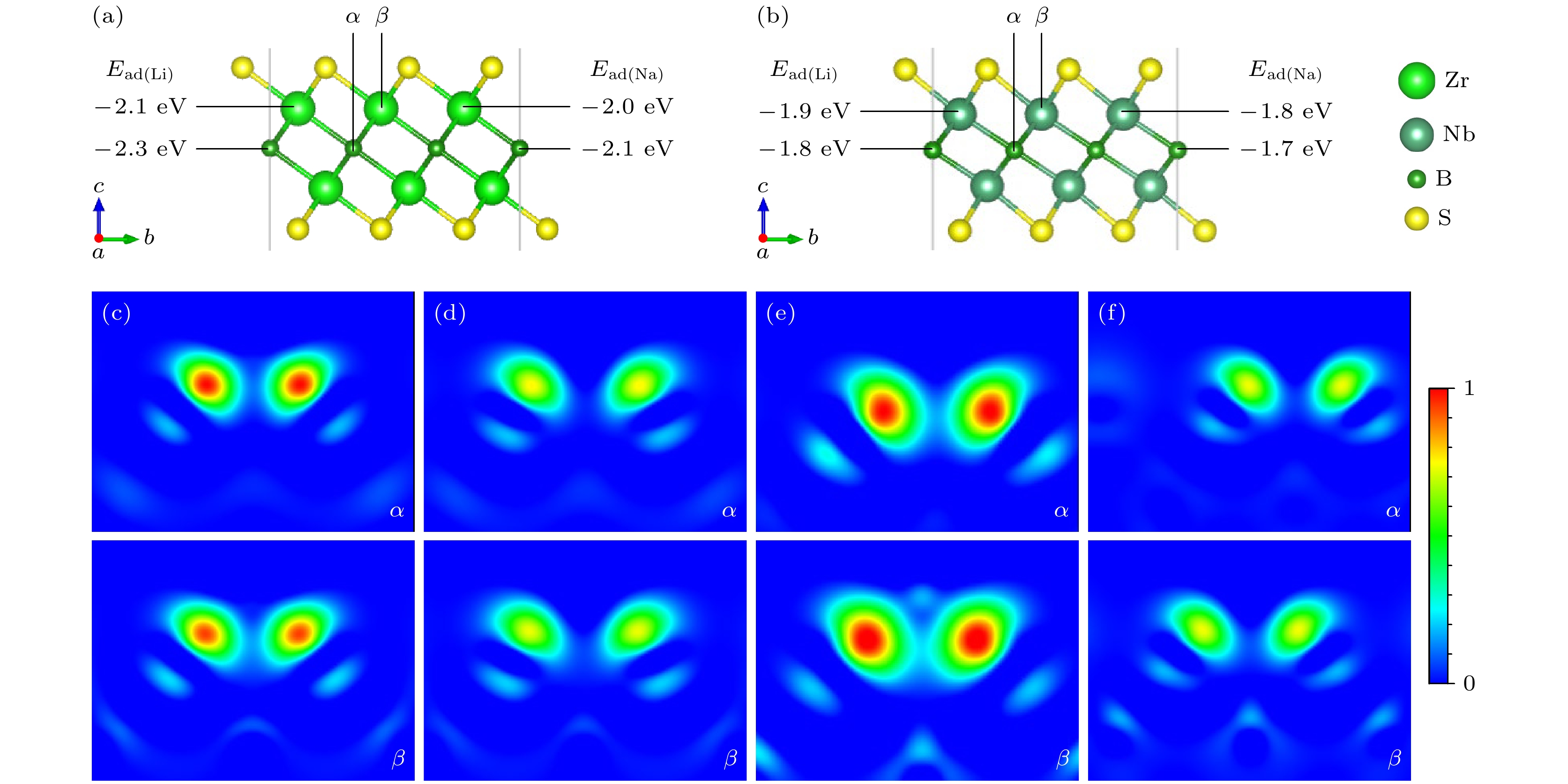
图 4 (a) Zr2BS2和(b) Nb2BS2结构模型及不同位点的吸附能; Zr2BS2 (0 1 0)截面的(c) Li和(d) Na不同位点的差分电荷密度; Nb2BS2 (0 1 0)截面的(e) Li和(f) Na不同位点的差分电荷密度
Figure 4. Structural model of (a) Zr2BS2 and (b) Nb2BS2 with adsorption energies at different sites; differential charge density for Li and Na at different sites on the (0 1 0) plane of (c), (d) Zr2BS2 and (e), (f) Nb2BS2.
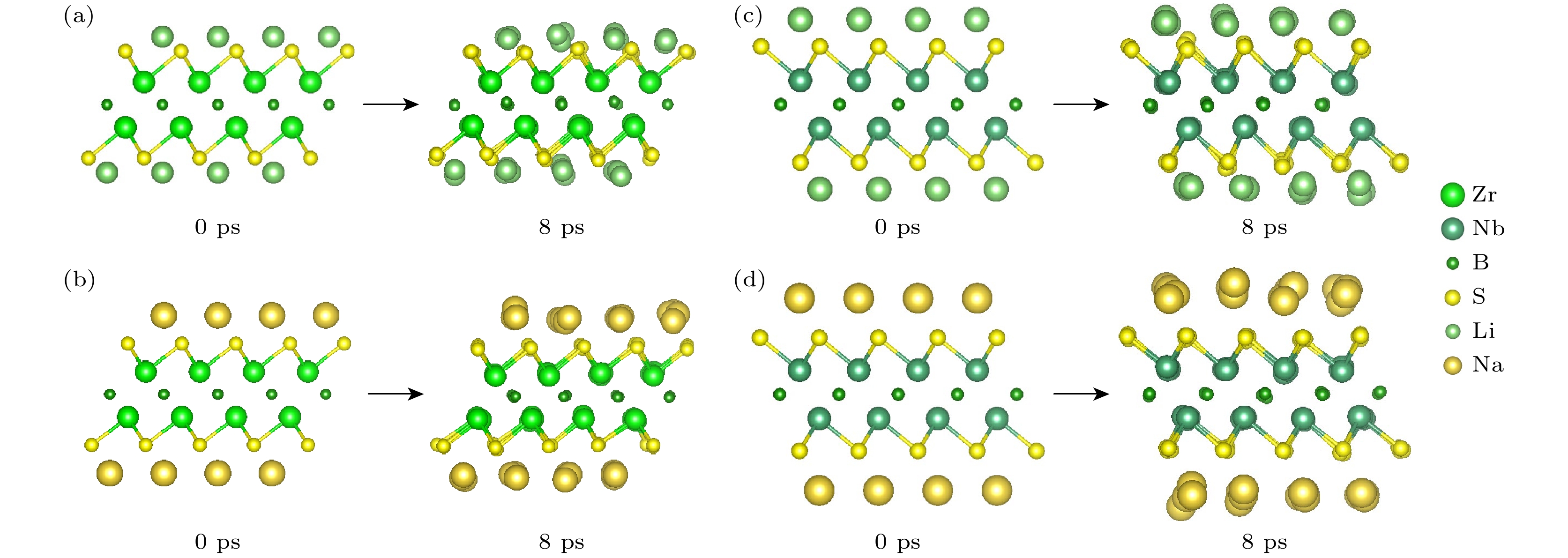
图 5 350 K下(a) Zr2BS2Li2, (b) Zr2BS2Na2, (c) Nb2BS2Li2和(d) Nb2BS2Na2在0 ps和8 ps时的AIMD模拟结构模型
Figure 5. AIMD-simulated structural models at 0 ps and 8 ps for (a) Zr2BS2Li2, (b) Zr2BS2Na2, (c) Nb2BS2Li2, and (d) Nb2BS2Na2 at 350 K.
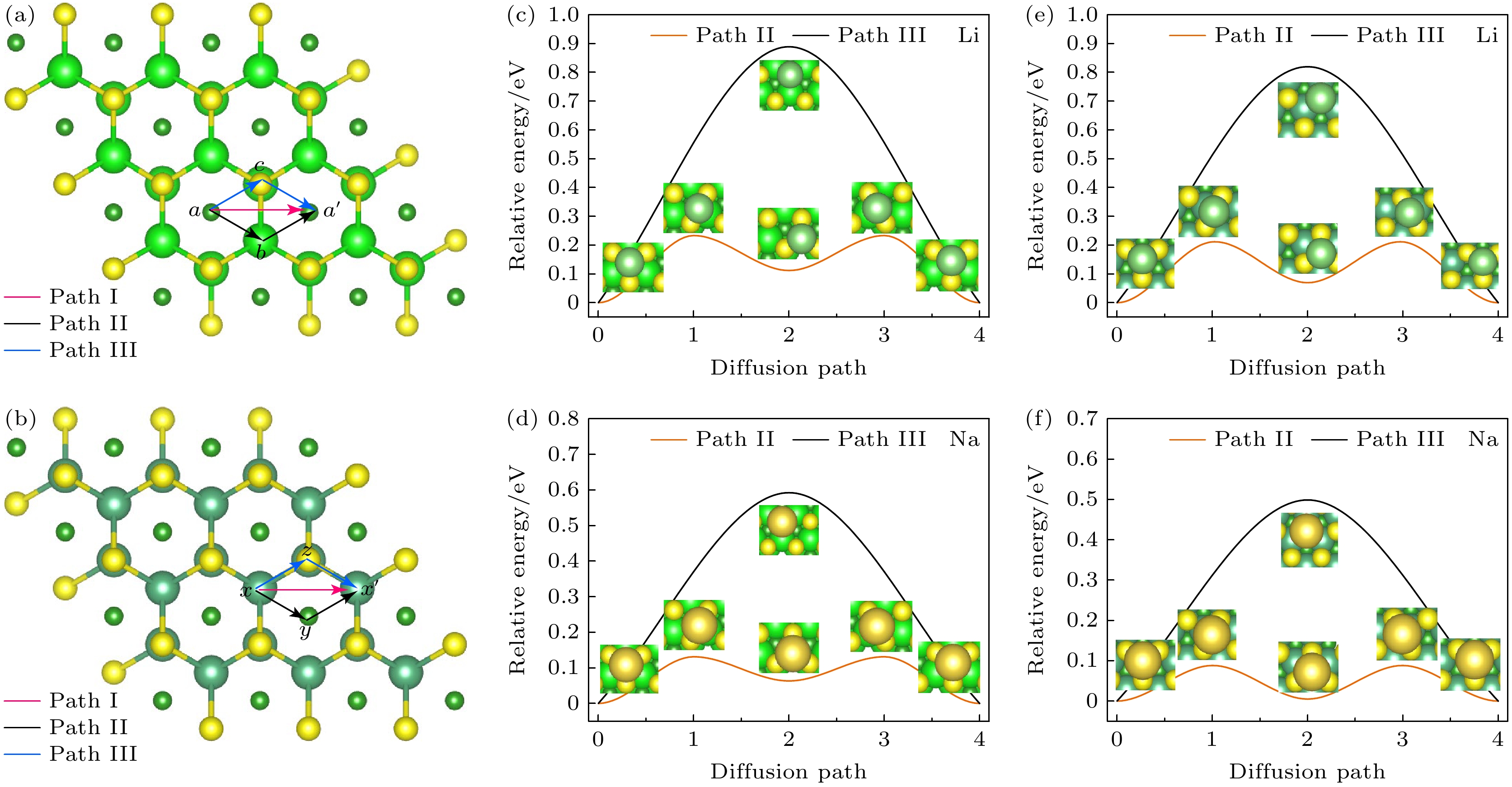
图 6 (a) Zr2BS2和(b) Nb2BS2表面扩散路径; Zr2BS2表面(c) Li和(d) Na扩散势垒; Nb2BS2表面(e) Li和(f) Na扩散势垒
Figure 6. Surface diffusion paths of (a) Zr2BS2 and (b) Nb2BS2; diffusion barriers for Li and Na on the surface of (c), (d) Zr2BS2 and (e), (f) Nb2BS2.
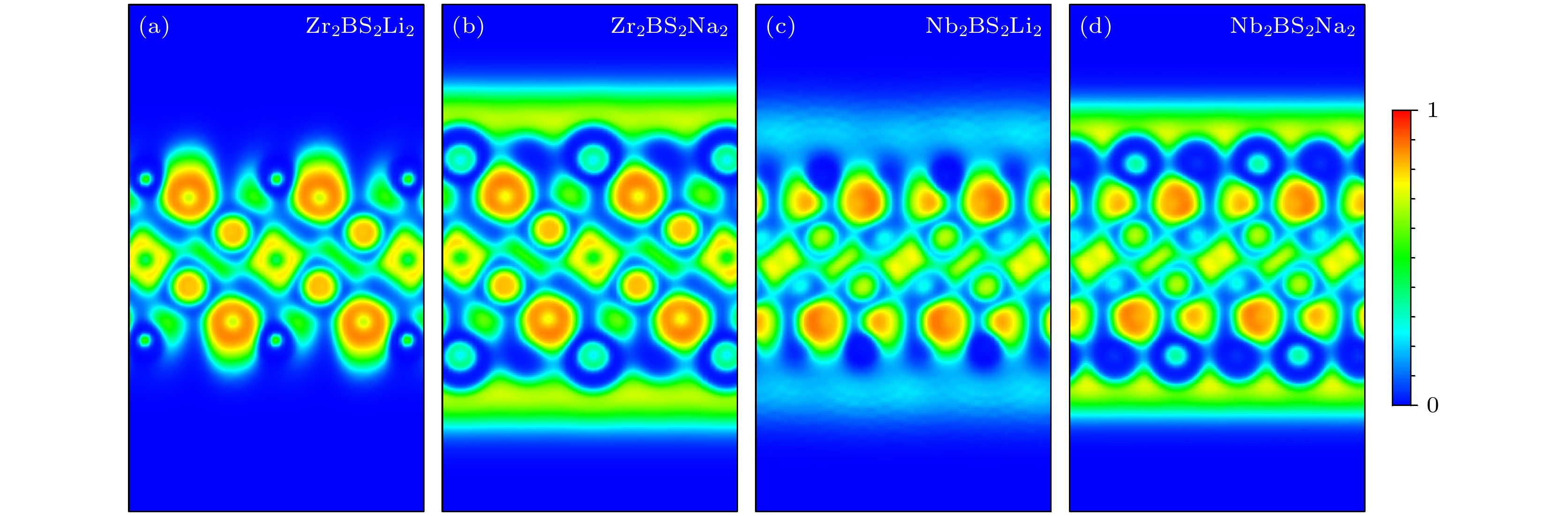
图 7 (1 1 0)截面的(a) Zr2BS2Li2, (b) Zr2BS2Na2, (c) Nb2BS2Li2和(d) Nb2BS2Na2电子局域函数
Figure 7. ELF on the (1 1 0) plane of (a) Zr2BS2Li2, (b) Zr2BS2Na2, (c) Nb2BS2Li2, and (d) Nb2BS2Na2.
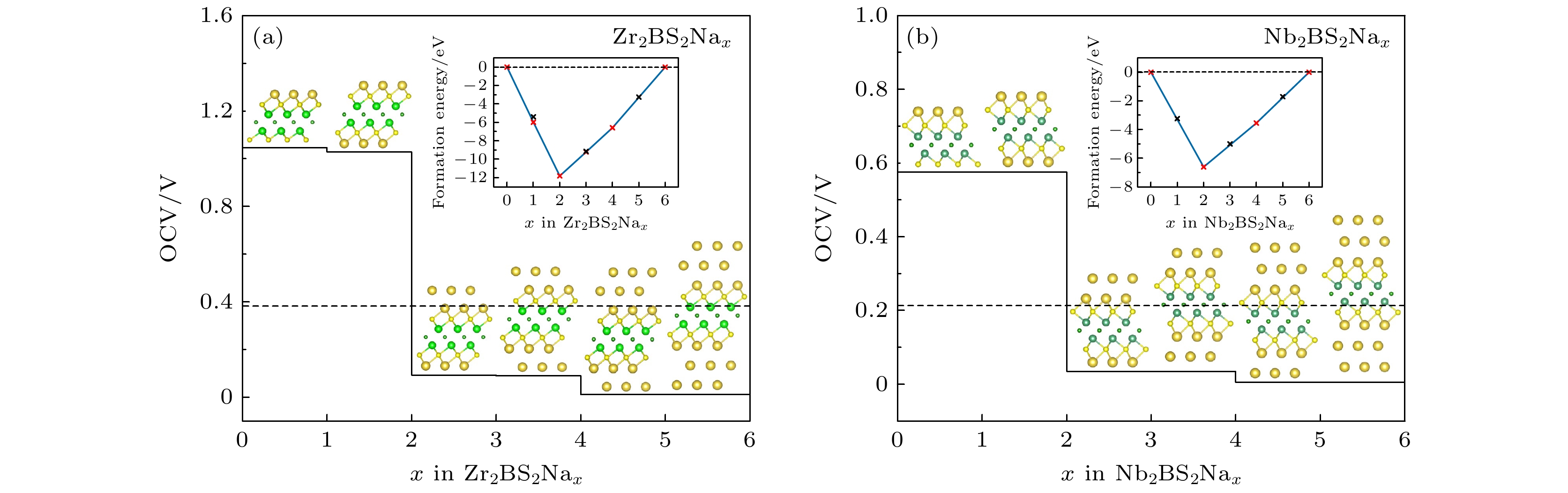
图 8 (a) Zr2BS2Nax和(b) Nb2BS2Nax开路电压
Figure 8. Open-circuit voltage of (a) Zr2BS2Nax and (b) Nb2BS2Nax.
表 1 Zr2BS2, Nb2BS2和MoS2的弹性常数、杨氏模量、泊松比计算结果及文献[38]结果
Table 1. Calculated results of elastic constants, Young’s modulus, Poisson’s ratio of Zr2BS2, Nb2BS2, and MoS2, and the results from Ref. [38].
System C11/
(N·m–1)C22/
(N·m–1)C12/
(N·m–1)$ E_{x(y)}^{\rm 2D} $ /
(N·m–1)$ v_{x(y)}^{2{\text{D}}} $ Zr2BS2 131.1 131.1 28.1 125.1 0.21 Nb2BS2 197.2 197.2 43.0 187.8 0.22 MoS2 135.4 135.4 30.6 128.4 0.23 MoS2[38] 132.3 132.3 32.8 124.1 0.25  下载: 导出CSV
下载: 导出CSV
-
[1] Dusastre V, Martiradonna L 2017 Nat. Mater. 16 15 doi: 10.1038/nmat4838 [2] Tian J J, Xue Q F, Yao Q, Li N, Christoph J, Brabec, Hin L Y 2020 Adv. Energy Mater. 10 2000183 doi: 10.1002/aenm.202000183 [3] Akkerman Q A, Gandini M, Stasio F D, Rastogi P, Palazon F, Bertoni G, Ball J M, Prato M, Petrozza A, Manna L 2016 Nat. Energy 2 16194 doi: 10.1038/nenergy.2016.194 [4] Barre A, Deguilhem B, Grolleau S, Gérard M, Suard F, Riu D 2013 J. Power Sources 241 680 doi: 10.1016/j.jpowsour.2013.05.040 [5] Wang Y X, Liu B, Li Q Y, Cartmell S, Ferrara S, Deng Z Q, Xiao J 2015 J. Power Sources 286 330 doi: 10.1016/j.jpowsour.2015.03.164 [6] Jin L M, Shen C, Shellikeri A, Wu Q, Zheng J S, Andrei P, Zhang J G, Zheng J P 2020 Energy Environ. Sci. 13 2341 doi: 10.1039/D0EE00807A [7] Noori A, El-Kady M F, Rahmanifar M S, Kaner R B, Mousavi M F 2019 Chem. Soc. Rev. 48 1272 doi: 10.1039/C8CS00581H [8] Soltani M, Beheshti S H 2021 J. Energy Storage 34 102019 doi: 10.1016/j.est.2020.102019 [9] Choi N S, Chen Z, Freunberger S A, Ji X, Sun Y K, Amine K, Yushin G, Nazar L F, Cho J, Bruce P G 2012 Angew. Chem. Int. Ed. 51 9994 doi: 10.1002/anie.201201429 [10] Fang Y J, Xiao L F, Chen Z X, Ai X P, Cao Y L, Yang H X 2018 Electrochem. Energy Rev. 1 294 doi: 10.1007/s41918-018-0008-x [11] Li F, Tang Q 2019 ACS Appl. Nano Mater. 2 7220 doi: 10.1021/acsanm.9b01718 [12] Zhang B K, Zhou J, Guo Z L, Peng Q, Sun Z M 2020 Appl. Surf. Sci. 500 144248 doi: 10.1016/j.apsusc.2019.144248 [13] Liu X, Ge X L, Dong Y, Fu K, Meng F B, Si R H, Zhang M H, Xu X W 2020 Mater. Chem. Phys. 253 123334 doi: 10.1016/j.matchemphys.2020.123334 [14] Zhang B K, Zhou J, Sun Z M 2022 J. Mater. Chem. A 10 15865 doi: 10.1039/D2TA03482D [15] Guo Z L, Zhou J, Sun Z M 2017 J. Mater. Chem. A 5 23530 doi: 10.1039/C7TA08665B [16] Zha X H, Xu P, Huang Q, Du S, Zhang R Q 2020 Nanoscale Adv. 2 347 doi: 10.1039/C9NA00610A [17] Ma N G, Wang T R, Li N, Li Y R, Fan J 2022 Appl. Surf. Sci. 571 151275 doi: 10.1016/j.apsusc.2021.151275 [18] Jia J, Li B J, Duan S Q, Cui Z, Gao H T 2019 Nanoscale 11 20307 doi: 10.1039/C9NR05708K [19] Zhou J, Palisaitis J, Halim J, Dahlqvist M, Tao Q, Persson I, Hultman L, Persson P O Å, Rosen J 2021 Science 373 801 doi: 10.1126/science.abf6239 [20] Khaledialidusti R, Khazaei M, Wang V, Miao N, Si C, Wang J, Wang J 2021 J. Phys.: Condens. Matter 33 155503 doi: 10.1088/1361-648X/abbb0e [21] Liang B C, Ma N G, Wang Y H, Wang T R, Fan J 2022 Appl. Surf. Sci. 599 153927 doi: 10.1016/j.apsusc.2022.153927 [22] Mehta V, Saini H S, Srivastava S, Kashyap M K, Tankeshwar K 2019 J. Phys. Chem. C 123 25052 doi: 10.1021/acs.jpcc.9b05679 [23] Li D Q, Chen X F, Xiang P, Du H Y, Xiao B B 2020 Appl. Surf. Sci. 501 144221 doi: 10.1016/j.apsusc.2019.144221 [24] Wang Y H, Ma N G, Liang B C, Fan J 2022 Appl. Surf. Sci. 596 153619 doi: 10.1016/j.apsusc.2022.153619 [25] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169 doi: 10.1103/PhysRevB.54.11169 [26] Kresse G, Furthmüller J 1996 Comput. Mater. Sci. 6 15 doi: 10.1016/0927-0256(96)00008-0 [27] Blöchl P E 1994 Phys. Rev. B 50 17953 doi: 10.1103/PhysRevB.50.17953 [28] Kresse G, Joubert D 1999 Phys. Rev. B 59 1758 doi: 10.1103/PhysRevB.59.1758 [29] Perdew J P, Ernzerhof M, Burke K 1996 J. Chem. Phys. 105 9982 doi: 10.1063/1.472933 [30] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 doi: 10.1103/PhysRevLett.77.3865 [31] Grimme S 2006 J. Comput. Chem. 27 1787 doi: 10.1002/jcc.20495 [32] Gonze X, Lee C 1997 Phys. Rev. B 55 10355 doi: 10.1103/PhysRevB.55.10355 [33] Togo A, Tanaka I 2015 Scr. Mater. 108 1 doi: 10.1016/j.scriptamat.2015.07.021 [34] Paier J, Hirschl R, Marsman M, Kresse G 2005 J. Chem. Phys. 122 234102 doi: 10.1063/1.1926272 [35] Yuan X, Zhang Z Y, He Y P, Zhao S Q, Zhou N G 2022 J. Phys. Chem. C 126 91 doi: 10.1021/acs.jpcc.1c09237 [36] Yan B Z, Lu C J, Zhang P G, Chen J, He W, Tian W B, Zhang W, Sun Z M 2020 Mater. Today Commun. 22 100713 doi: 10.1016/j.mtcomm.2019.100713 [37] Chen Z H, Huang S W, Yuan X, Gan X L, Zhou N G 2021 Appl. Surf. Sci. 544 148861 doi: 10.1016/j.apsusc.2020.148861 [38] Singh S, Espejo C, Romero A H 2018 Phys. Rev. B 98 155309 doi: 10.1103/PhysRevB.98.155309 [39] Andrew R C, Mapasha R E, Ukpong A M, Chetty N 2012 Phys. Rev. B 85 125428 doi: 10.1103/PhysRevB.85.125428 [40] Born M, Huang K 1996 Dynamical Theory of Crystal Lattices (New York: Oxford University Press) pp129–165 [41] Shu H B, Li F, Hu C L, Liang P, Cao D, Chen X S 2016 Nanoscale 13 2918 doi: 10.1039/C5NR07909H [42] Zhang X M, Yu Z M, Wang S S, Guan S, Yang H Y, Yao Y G, Yang S A 2016 J. Mater. Chem. A 4 15224 doi: 10.1039/C6TA07065E [43] Meng Q Q, Ma J L, Zhang Y H, Li Z, Hu A, Kai J J, Fan J 2018 J. Mater. Chem. A 6 13652 doi: 10.1039/C8TA04417A [44] Meng Q Q, Ma J L, Zhang Y H, Li Z, Zhi C Y, Hu A, Fan J 2018 Nanoscale 10 3385 doi: 10.1039/C7NR07649E [45] Shukla V, Jena N K, Naqvi S R, Luo W, Ahuja R 2019 Nano Energy 58 877 doi: 10.1016/j.nanoen.2019.02.007 [46] Gao S L, Hao J B, Zhang X H, Li L, Zhang C L, Wu L Y, Ma X G, Lu P F, Liu G 2021 Comput. Mater. Sci. 200 110776 doi: 10.1016/j.commatsci.2021.110776 [47] Urban A, Seo D H, Ceder G 2016 npj Comput. Mater. 2 16002 doi: 10.1038/npjcompumats.2016.2 [48] Aydinol M K, Kohan A F, Ceder G, Cho K, Joannopoulos J 1997 Phys. Rev. B 56 1354 doi: 10.1103/PhysRevB.56.1354 [49] Eames C, Islam M S 2014 J. Am. Chem. Soc. 136 16270 doi: 10.1021/ja508154e [50] Yang E, Ji H, Kim J, Kim H, Jung Y 2015 Phys. Chem. Chem. Phys. 17 5000 doi: 10.1039/C4CP05140H -
计量
- 文章访问数: 173
- HTML全文浏览数: 173
- PDF下载数: 6
- 施引文献: 0