
 首页
首页 登录
登录 注册
注册



-
新精神活性物质(new psychoactive substances,NPS)又称“策划药”或“合法毒品”[1],联合国毒品与犯罪办公室(The United Nations Office for Drugs and Crime, UNODC)定义其为不受《1961年麻醉品单一公约》或《1971年精神药物公约》约束,可能危害公共健康的纯品或制剂形式的滥用物质[2]。NPS是继传统毒品、合成毒品后全球流行的第三代毒品,已成为全球性的健康和社会问题[3]。
质谱法是检测NPS的有力工具,被认为是鉴定已知化合物的有效传统策略[4]。通常通过算法将待测化合物质谱图与谱库中已有的标准物质谱图进行相似性搜索来识别化合物。但该策略只能识别数据库中已有的化合物,而不法分子为了逃避监管,会对化合物结构进行修饰改造,给基于参比物质或质谱数据库的传统技术筛查和物质鉴定带来了挑战。
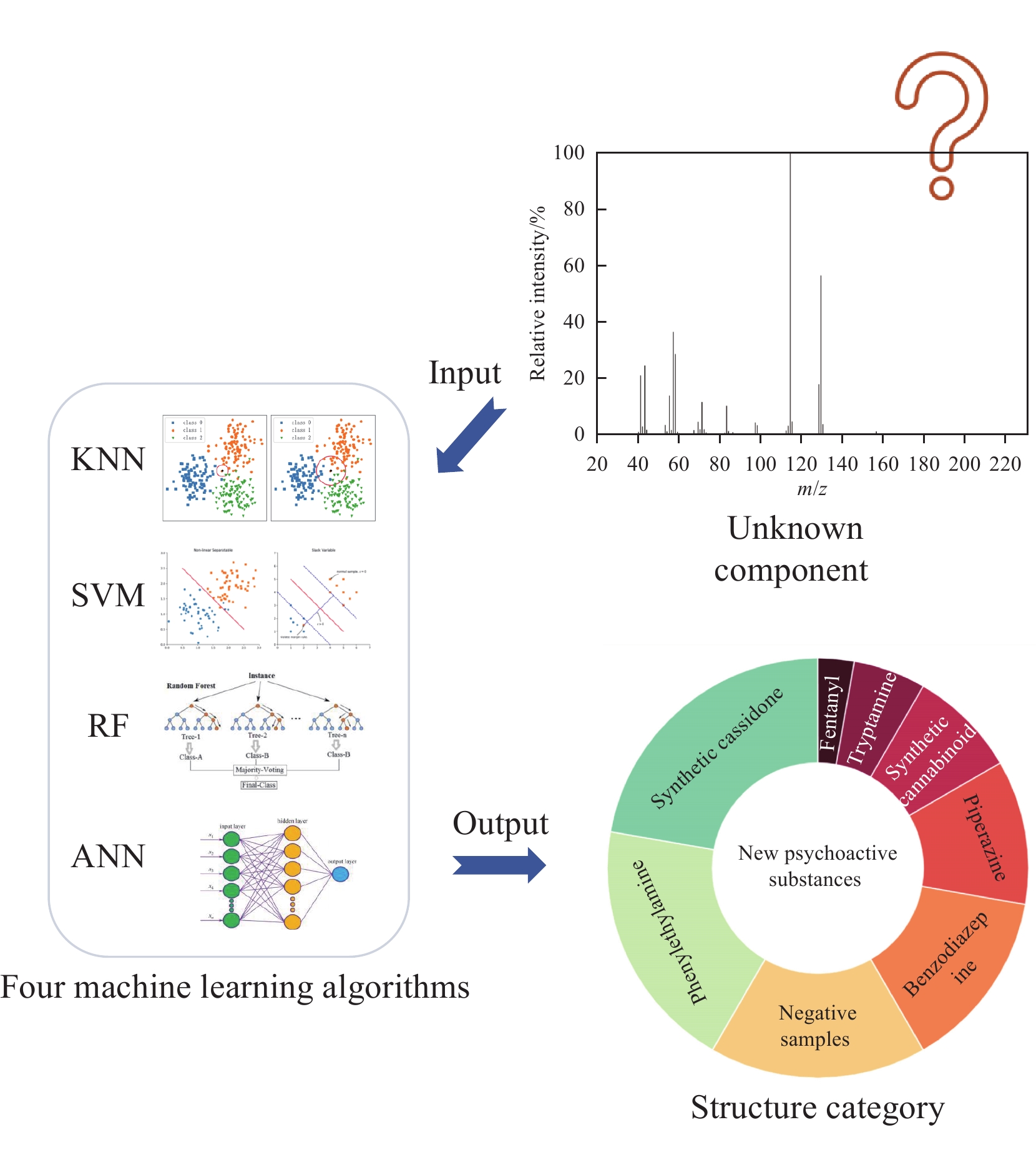
机器学习的进步已成为解决这一问题的潜在方法。Koshute等[5]提出有监督的机器学习分类模型用于辅助质谱检测芬太尼类似物,实现了99%检测准确率,证明了机器学习模型可以为数据库匹配提供强大的补充。Yan等[6]使用包含567个LC-MS 和732个GC-MS数据的数据集生成并评估了K近邻、支持向量机、随机森林和多粒度级联森林等4种分类模型快速筛查新精神活性物质。Lee等[7]构建了基于高分辨液相色谱-串联质谱的机器学习模型,以解决识别已列管物质和未知新型精神活性物质的分析挑战。但这些方法覆盖的NPS种类和范围有限,缺乏基于电子电离质谱(electron ionization mass spectrometry,EI-MS)对NPS较为全面的分类模型构建。
本研究将基于7种NPS的EI-MS数据,建立最近邻算法(k-nearest neighbor, KNN)、支持向量机(support vector machine, SVM)、随机森林(random forests, RF)、人工神经网络(artificial neural network, ANN)4种分类机器学习模型,以实现仅使用EI-MS数据识别未知NPS,为未知NPS的结构鉴定提供新策略。
-
本研究收集了671个NPS和200个阴性样本的EI-MS数据,这些数据来自于871个不同的样本,为缉获药物分析科学工作组数据库(The Scientific Working Group for the Analysis of Seized Drugs, SWGDRUG)以及实验室对毒品标准品进行EI-MS分析得到的数据,将其保存为.CSV格式,逐峰导出m/z 41~400碎片离子丰度,并归一化到1。按照化学结构对化学物质进行分类并赋予相应的标签,0代表合成卡西酮类物质,共包含89个样本;1代表苯乙胺类物质,共包含112个样本;2代表哌嗪类物质,共包含50个样本;3代表色胺类物质,共包含67个样本;4代表芬太尼类物质,共包含222个样本;5代表合成大麻素类物质,共包含70个样本;6代表苯二氮卓类物质,共包含62个样本;7代表阴性样本,共包含200个样本。
-
Agilent MS5975质谱仪:美国Agilent公司产品,电子电离模式,电离能70 eV,离子源温度200 ℃,传输线温度180 ℃,SCAN模式,质量扫描范围m/z 41~400。
-
机器学习模型的构建以及优化均使用python(版本 3.10, 2021)语言,使用train_test_split函数将数据集划分为训练集和测试集,比例为7∶3。在训练集上调用fit函数进行模型训练,在测试集上评估模型的泛化能力。使用RF、KNN、SVM、ANN等4种有监督的机器学习模型对所搜集的数据集进行训练,在KNN分析前,先对数据进行降维处理,采用常用的降维方法主成分分析,通过sklearn的Dimensionality reduction模块中PCA函数完成。代码参见github链接
https://github.com/xufeizhai/A-Classification-and-Prediction-Model-for-New-Psychoactive-Substances 。 -
在绝大多数非线性模型中,有一部分参数是无法通过训练直接获取的,通常的做法是直接预先设定,再反复调整使模型达到最优状态,这一过程即调参过程。本研究使用5倍交叉验证的网格搜索进行优化,以获得每个模型的最佳学习超参数。将训练集均匀划分为5个互不重叠的子集,在每一次迭代中,选取其中的4个子集合并作为当前的训练集,而剩余的1个子集用作验证集,以评估模型的性能。这个过程循环进行,确保每个子集都有机会作为验证集使用。当所有子集都依次作为验证集完成1轮评估后,计算模型在所有子集中性能的平均值,以此作为模型整体性能的度量。在训练子集中,通过网格搜索遍历参数,每个模型的最优超参数列于表1。该过程是借助sklearn中GridSearchCV模块实现的,会尝试本研究所关心参数的所有可能组合,通常考虑以下几个参数:Estimator被调参数的模型;Param_grid被调参数的grid;使用Scoring函数作为模型的评价指标;CV一般直接取整数即可,即k-fold中的k,设置为5。
-
本研究采用混淆矩阵以及4个多分类模型的常见评价指标,即准确率(accuracy)、精确度(precision)、召回率(recall)、f-分数(F1 score)对模型性能进行评价,计算方法示于式(1)~(4)。
混淆矩阵是机器学习中总结分类模型预测结果的情形分析表,以矩阵形式将数据集中的样本按照真实的类别与分类模型预测的类别进行汇总。其中,矩阵的行和列分别表示真实值和预测值。混淆矩阵中4个参数的说明情况示于图1。其中,真阳性率(true positive,TP)指真实值是positive,模型认为是positive的数量;假阴性率(false negative,FN)指真实值是positive,模型认为是negative的数量;假阳性率(false positive,FP)指真实值是negative,模型认为是positive的数量;真阴性率(true negative,TN)指真实值是negative,模型认为是negative的数量。
针对整个模型,准确度指分类模型所有判断正确的结果占总观测值的比值;精确度指在模型预测是positive的所有结果中,模型预测正确的比值;灵敏度即召回率,指在真实值是positive的所有结果中,模型预测正确的比值。利用classification_report函数可以计算每个类的准确度、精确度、召回率和f-分数,代码参见github链接,见1.3.1节。
-
本研究基于671个NPS和200个阴性样本的EI-MS数据,构建了KNN、SVM、RF、ANN等4种分类预测模型。通过5折交叉验证法对各模型进行超参数调整,获得各模型的最优超参数,以提升模型的分类准确度。采用EI-MS数据集测试集中的261个样本,其中,包括29份卡西酮类、30份苯乙胺类、18份哌嗪类、16份色胺类、64份色胺类、24份合成大麻素类、17份苯二氮卓类、63份阴性样本对模型进行评价。各个模型的混淆矩阵示于图2。在单个混淆矩阵中,所有数字的总和表示测试集的数量,其中,每一行代表真实样本的预测结果(召回率),每一列代表预测结果的实际类别(精度)。模型的评价结果列于表2,展示了4种模型对8种类型样本的识别能力。
ANN、SVM、RF等3种算法具有处理高维数据的能力,不对数据集进行PCA降维,可以更大程度地保留质谱数据信息,以获得更好的分类效果;KNN算法对高维数据的处理效果不佳,需首先进行PCA降维处理,但该算法简单、易实现。总体上,对于7种NPS以及阴性样本的分类预测效果,RF预测模型最好,整体准确度为89.27%,KNN、SVM、ANN模型的整体准确度分别为79.31%、83.14%、83.52%。另外,RF预测模型对具体类别的NPS预测具有较高的精确度,对合成卡西酮类、芬太尼类、合成大麻素类、苯二氮卓类的精确度分别为100%、93%、95%、100%,召回率分别为76%、100%、88%、100%,均高于其他3种模型。在使用KNN模型分类前,先对PCA数据集进行处理,保留14个主成分,将原有的360个特征降低至14个。该模型对于8类化合物整体的分类预测准确度低于80%,可能是由于输入的EI-MS数据集为大部分特征取值为0的稀疏数据集,KNN算法并不适用于这一数据集的分类预测,尤其是对哌嗪类物质进行预测时,在预测的18个哌嗪类物质中,仅有6个预测正确。
在RF模型矩阵中,第1、3、5、6、7、8列分别表明合成卡西酮、哌嗪、芬太尼、合成大麻素、苯二氮卓类似物以及阴性样本的精度分别为100.00%、100.00%、93.00%、95.0%、100.00%、86.00%,均高于85%。第2列表明,在被预测的36个苯乙胺类样本中,有3个样本被错误预测为卡西酮类物质,有3个样本被错误预测为哌嗪类物质,有3个样本被错误预测为阴性样本。在第4列被预测的19个色胺类样本中,有4个样本被错误预测为哌嗪类物质。值得注意的是,该模型对芬太尼类和苯二氮卓类物质的召回率均为100.00%。RF、SVM、ANN模型对合成大麻素类和芬太尼类物质的准确度均较高,分别为95%、95%、91%和93%、91%、89%。
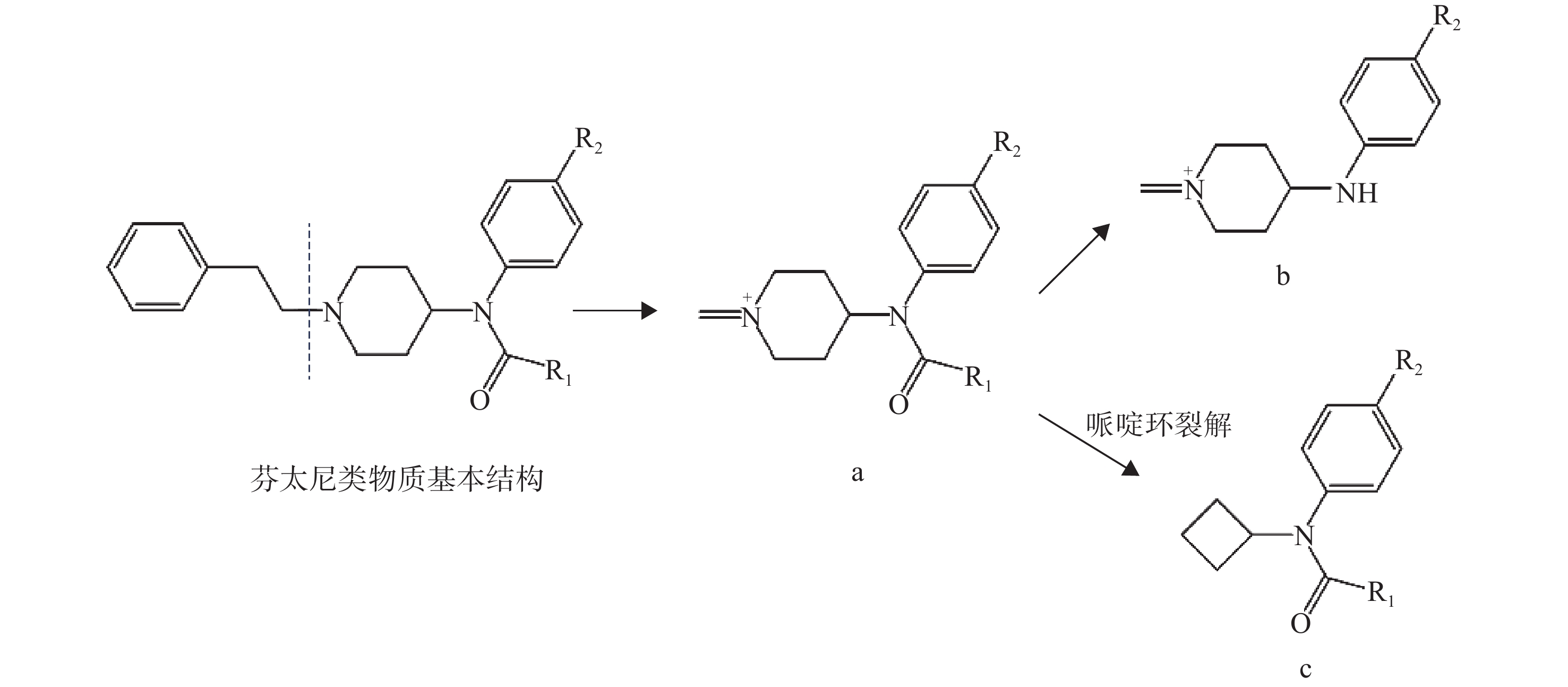
同类化合物往往具有相同的骨架和类似的分子结构,因而它们的质谱碎裂途径和主要的碎片离子具有高度的相似性,这是机器学习模型基于质谱数据对未知化合物进行分类预测的基础。合成卡西酮类物质存在β-羰基苯乙胺骨架结构,其优势碎片离子一般由羰基或C−N键断裂和苯环典型断裂产生。苯乙胺类物质的结构特异性较低,大多数通过C−N键断裂和苯环典型断裂产生碎片离子m/z 91,因此,苯乙胺类物质的识别具有挑战性。从结果来看,4个分类预测模型对苯乙胺类物质的识别精确度比其他类别NPS低。在EI模式下,芬太尼类物质哌啶环的裂解及哌啶环与苯乙基的断裂为主要的碎裂途径,哌啶环N原子失去π电子形成游离基中心,诱导相邻碳原子发生α断裂而丢失卓鎓离子形成特征离子a,进一步发生N-苯基酰胺解离,羰基与氨基之间化学键发生断裂得到碎片离子b,哌啶环裂解还会生成特征离子c,示于图3。合成大麻素类物质的结构母核一般为吲唑环或吲哚环,吲哚/吲唑N原子端侧链的不同会产生不同的特征离子,主要是羰基不饱和杂原子发生α断裂产生带有侧链基团的吲唑/吲哚酰鎓离子。通过电子轰击苯二氮卓类物质易产生具有苯基正离子的特征碎片离子m/z 283。另外,哌嗪类和色胺类物质不易区分,可能是由于这2类物质易产生相似的碎片离子,其结构示于图4。色胺类和哌嗪类物质均会产生m/z 174碎片离子,另外,还会分别产生m/z 145、146碎片离子。朱娜等[8]研究表明,苄基哌嗪类和苯基哌嗪类化合物在裂解过程中分别会产生m/z 146、174碎片离子。当色胺类物质的苯环上为甲氧基取代[9]时,该色胺类物质会首先失去1个电子得到分子离子,分子离子中的 C−N键发生α断裂产生碎片离子A(m/z 174),该分子离子发生β断裂得到碎片离子B,当R1为甲氧基时,碎片离子B中的CH3−O发生均裂,失去−CH3得到离子C(m/z 145),裂解途径示于图5。除此之外,用于数据训练的哌嗪类和色胺类物质的样本数量分别为50和67,由于样本量较少,模型较难学习识别化合物的结构特征,因此分类预测模型不易区分这2类物质。
-
本研究基于7种NPS的EI-MS数据,使用KNN、RF、SVM、ANN 4个机器学习模型构建分类模型,对超参数进行优化,用准确率、精确度、召回率、f1-分数等指标评估。结果表明,RF模型对7种NPS的分类预测性能最好,整体准确度为89.27%,可以很好地预测未知化合物的结构类别。本文提供了一种利用机器学习算法快速解析新精神活性物质的策略,能够实现对未知化合物结构类别的分类预测,为其结构鉴定提供依据。
基于电子电离质谱数据和机器学习的新精神活性物质分类预测模型构建
Construction of Prediction Models for Classification of New Psychoactive Substances Based on EI-MS Data and Machine Learning
-
摘要: 新精神活性物质的结构变化快速,给基于标准物质和质谱数据库筛选和鉴定这些新物质带来了挑战。本研究使用机器学习方法为未知新精神活性物质的结构鉴定提供新策略。基于871个质谱数据集构建了最近邻、支持向量机、随机森林和人工神经网络算法用于新精神活性物质的结构分类预测,采用5倍交叉验证的网格搜索对模型的超参数进行优化,使用混淆矩阵、准确度、精密度、召回率和f-分数评估4种分类预测模型的性能。结果表明,随机森林模型的预测能力最优,整体准确度可达89.27%,可以很好地对未知化合物结构类别进行预测,从而为未知化合物的结构鉴定提供依据。
-
关键词:
- 电子电离质谱(EI-MS) /
- 新精神活性物质 /
- 机器学习 /
- 分类预测模型
Abstract: New psychoactive substances (NPS) have become a global health and social problem. Their structures are variable and can be easily modified to produce new compounds. Traditional analytical techniques mostly rely on standard substances and mass spectrometry databases. The increased structural diversity of NPS makes the mass spectrometry databases be unable to comprehensively cover the mass spectra of all possible NPS, which in turn makes it difficult to perform structural identification of completely unknown compounds. Advances in machine learning have emerged as a potential solution to this dilemma. In this study, the k-nearestneighbor (KNN), support vector machine (SVM), random forests (RF) and artificial neural network (ANN) algorithms were constructed based on a dataset of mass spectra of 871 compounds. The four algorithmic models for identifying new psychoactive substances were used for structural classification prediction. The training and test sets were divided according to the ratio of 7:3, and the fit method was invoked on the training set to construct the model and train the parameters of the model, and the generalization ability of the model was evaluated on the test set. A grid search with 5-fold cross-validation was used to optimize the hyperparameters of the models. The performance of the four classification prediction models was evaluated by using the confusion matrix, accuracy, precision, recall and f-scores for each of the four models for characterizing 261 samples from the test set. Overall, the RF prediction model has the best classification prediction for the seven NPS as well as negative samples, with an overall accuracy of 89.27%, which is higher than the other three classification prediction models. The overall accuracies of the KNN, SVM, and ANN models are 79.31%, 83.14%, and 83.52%, respectively. In addition, the RF prediction model also has high accuracy for the NPS prediction of specific classes, and the accuracies for synthetic cathinones, fentanyl, synthetic cannabinoids, and benzodiazepines are 100%, 93%, 95%, and 100%, respectively, which can warrant good prediction for the structural classes of unknown compounds. In conclusion, this study develops a strategy for rapid analysis of new psychoactive substances using machine learning algorithms based on mass spectral datasets, realizing the classification prediction of structural classes of unknown compounds, thus providing a basis for the structural identification of unknown psychoactive compounds. -

-
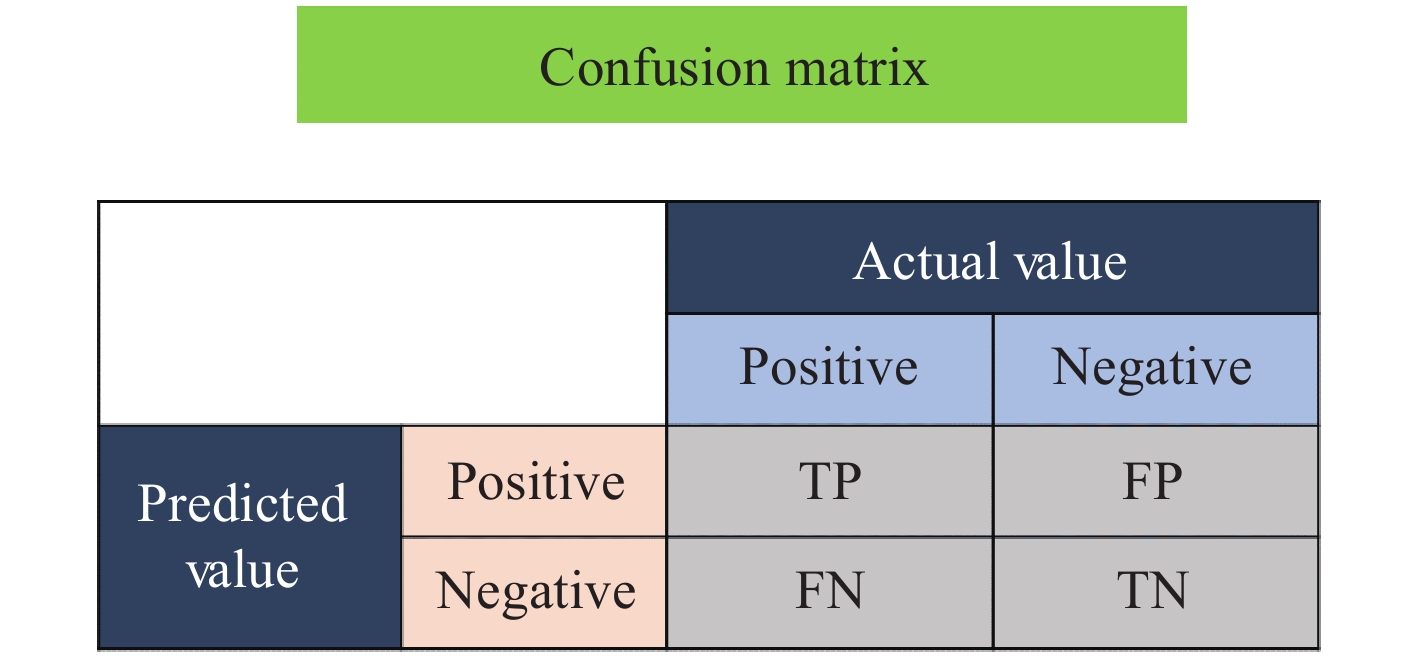
图 1 混淆矩阵中4个参数的说明
Figure 1. Illustration of the four parameters in the confusion matrix
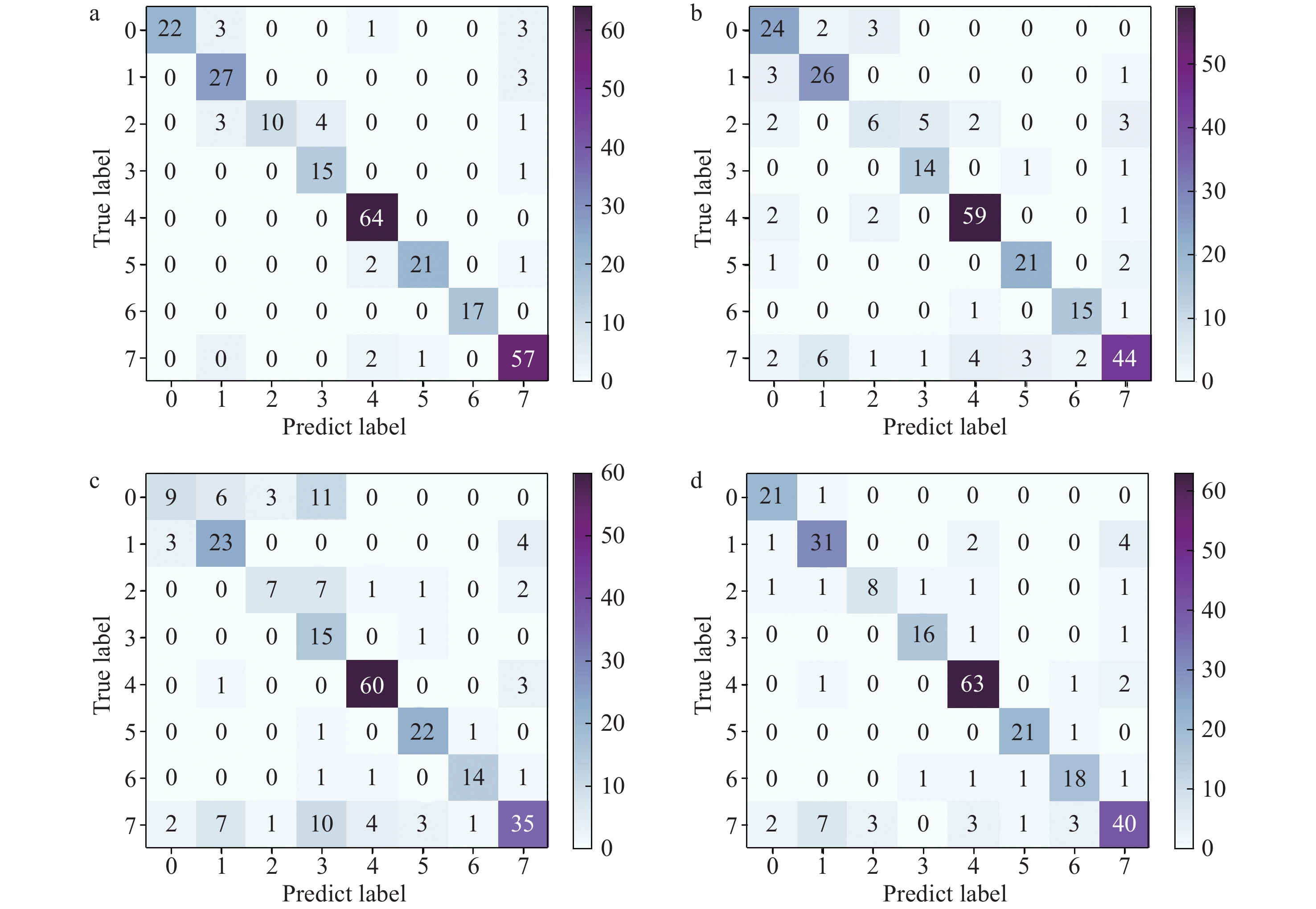
图 2 RF(a)、KNN(b)、SVM(c)、ANN(d)等4种模型的混淆矩阵
Figure 2. Confusion matrix of four models for RF (a), KNN (b), SVM (c) and ANN (d)
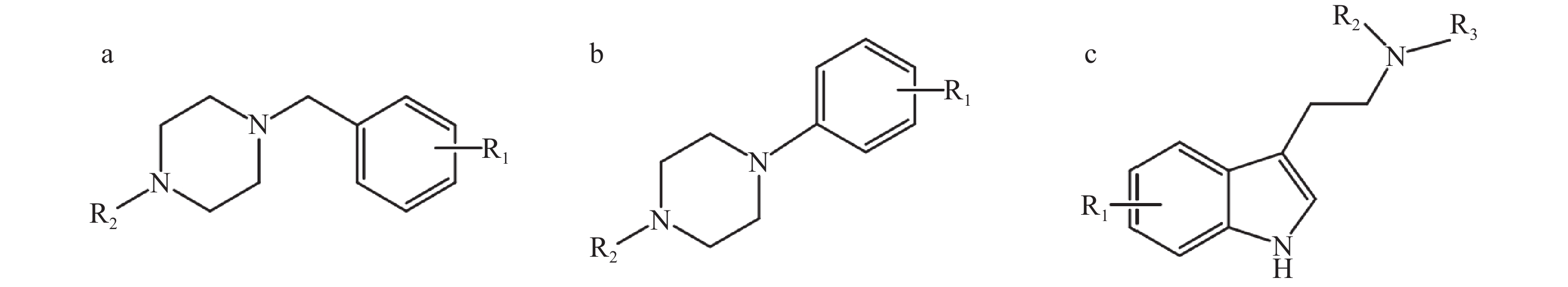
图 4 哌嗪类和色胺类新精神活性物质的结构
Figure 4. Structures of new psychoactive substances for piperazine and tryptamine
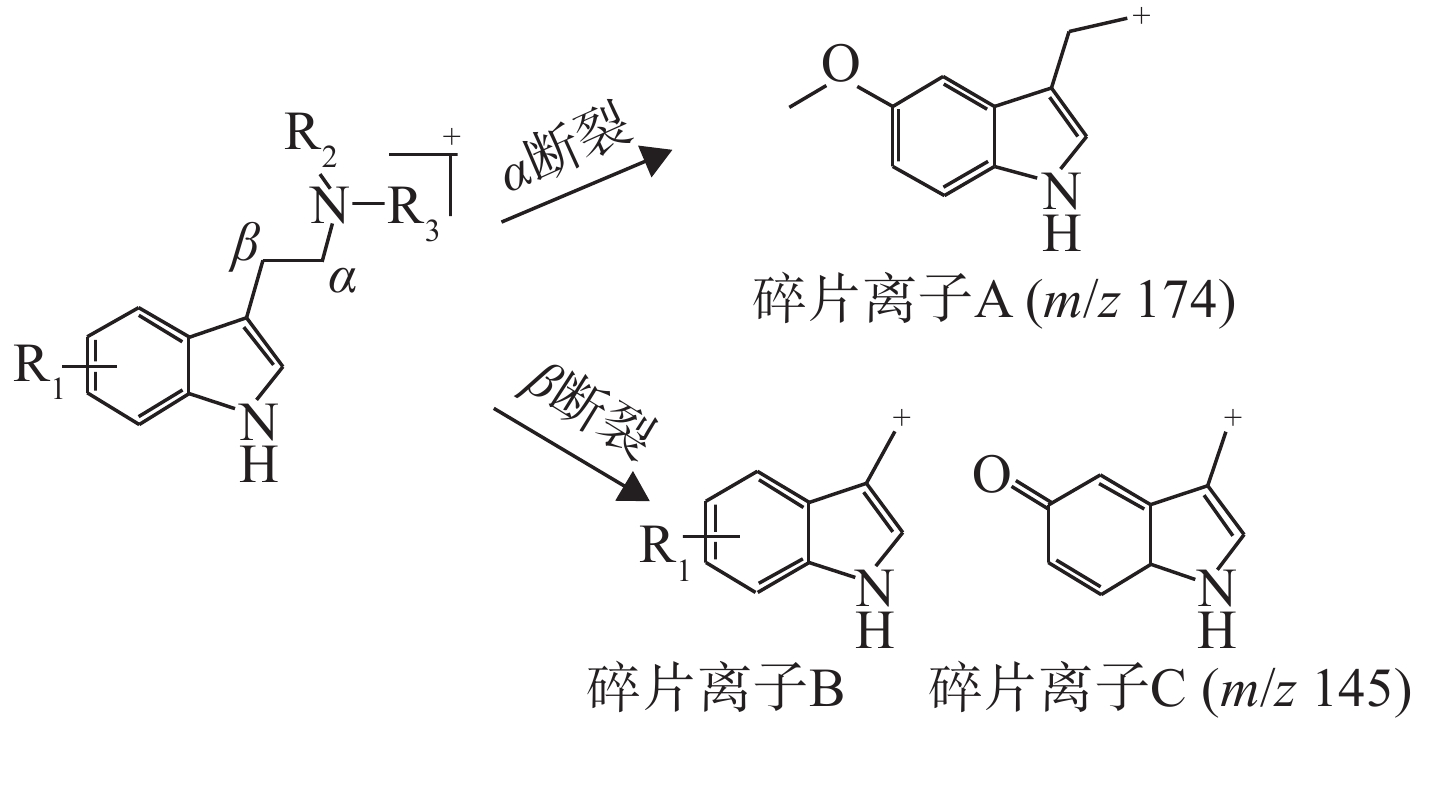
图 5 色胺类新精神活性物质的主要碎片离子结构
Figure 5. Structures of major fragment ions of new psychoactive substances for tryptamine
表 1 用于模型的各个超参数
Table 1. Hyperparameters used for models
模型名称 Model 超参数 Hyperparameter RF n_estimators=125 KNN n_neighbors=8;weights='distance';p=2 SVM Kernel = 'rbf';gamma=0.27;C=22.65 ANN hidden_layer_size s=(30,50);solver='adam';activation='relu';max_iter=3000  下载: 导出CSV
下载: 导出CSV
表 2 4种模型在8类化合物分类中的精确度、召回率和f-分数
Table 2. Precision, recall and f-score of the four models in the classification of 8 classes of compounds
模型
Model类别
Type精确度
Precision召回率
Recallf-分数
F1 scoreRF 合成卡西酮 1.00 0.76 0.86 苯乙胺 0.75 0.9 0.82 哌嗪 1.00 0.56 0.71 色胺 0.79 0.94 0.86 芬太尼 0.93 1.00 0.96 合成大麻素 0.95 0.88 0.91 苯二氮卓 1.00 1.00 1.00 阴性样本 0.86 0.90 0.88 KNN 合成卡西酮 0.69 0.83 0.75 苯乙胺 0.74 0.87 0.80 哌嗪 0.50 0.33 0.40 色胺 0.70 0.88 0.78 芬太尼 0.89 0.92 0.91 合成大麻素 0.81 0.88 0.84 苯二氮卓 0.94 0.88 0.91 阴性样本 0.82 0.67 0.74 SVM 合成卡西酮 0.72 0.82 0.77 苯乙胺 0.70 0.84 0.76 哌嗪 0.71 0.38 0.50 色胺 0.94 0.94 0.94 芬太尼 0.91 0.96 0.93 合成大麻素 0.95 0.86 0.90 苯二氮卓 0.84 0.73 0.78 阴性样本 0.82 0.78 0.80 ANN 合成卡西酮 0.84 0.95 0.89 苯乙胺 0.76 0.82 0.78 哌嗪 0.73 0.62 0.67 色胺 0.89 0.89 0.89 芬太尼 0.89 0.94 0.91 合成大麻素 0.91 0.95 0.93 苯二氮卓 0.78 0.82 0.80 阴性样本 0.82 0.68 0.74
下载: 导出CSV
-
[1] LUETHI D, LIECHTI M E. Designer drugs: mechanism of action and adverse effects[J]. Archives of Toxicology, 2020, 94(4): 1 085-1 133. [2] United Nations Office on Drugs and Crime (UNODC). World drug report 2023[EB/OL]. (2023-06-26) [2024-03-27]. https://www.unodc.org/unodc/en/data-and-analysis/world-drug-report-2023.html. [3] SHAFI A, BERRY A J, SUMNALL H, WOOD D M, TRACY D K. New psychoactive substances: a review and updates[J]. Therapeutic Advances in Psychopharmacology, 2020, doi: 10.1177/2045125320967197. [4] PASIN D, CAWLEY A, BIDNY S, FU S. Current applications of high-resolution mass spectrometry for the analysis of new psychoactive substances: a critical review[J]. Analytical and Bioanalytical Chemistry, 2017, 409(25): 5 821-5 836. [5] KOSHUTE P, HAGAN N, JAMESON N J. Machine learning model for detecting fentanyl analogs from mass spectra[J]. Forensic Chemistry, 2022, 27: 100 379. [6] YANG Y, LIU D, HUA Z, XU P, WANG Y, DI B, LIAO J, SU M. Machine learning-assisted rapid screening of four types of new psychoactive substances in drug seizures[J]. Journal of Chemical Information and Modeling, 2023, 63(3): 815 -825 . doi: 10.1021/acs.jcim.2c01342[7] LEE S Y, LEE S T, SUH S, KO B J, BIN OH H. Revealing unknown controlled substances and new psychoactive substances using high-resolution LC-MS-MS machine learning models and the hybrid similarity search algorithm[J]. Journal of Analytical Toxicology, 2022, 46(7): 732 -742 . doi: 10.1093/jat/bkab098[8] 朱娜, 俞晨, 花镇东, 徐鹏, 王优美, 狄斌, 苏梦翔. 哌嗪类新精神活性物质的质谱特征研究[J]. 质谱学报, 2021, 42(1): 1 -7 . doi: 10.7538/zpxb.2019.0161 ZHU Na, YU Chen, HUA Zhendong, XU Peng, WANG Youmei, DI Bin, SU Mengxiang. Mass fragmentation characteristics of piperazine analogues[J]. Journal of Chinese Mass Spectrometry Society, 2021, 42(1):1 -7 (in Chinese). doi: 10.7538/zpxb.2019.0161[9] 钱振华, 花镇东. 基于特征性离子快速筛查和识别色胺类新精神活性物质[J]. 质谱学报, 2021, 42(3): 197 -206 . doi: 10.7538/zpxb.2020.0036 QIAN Zhenhua, HUA Zhendong. Rapid screening and identification of tryptamines based on characteristic ions[J]. Journal of Chinese Mass Spectrometry Society, 2021, 42(3):197 -206 (in Chinese). doi: 10.7538/zpxb.2020.0036 -
计量
- 文章访问数: 644
- HTML全文浏览数: 644
- PDF下载数: 11
- 施引文献: 0