
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
近几十年来, 原子团簇已成为多学科交叉的研究热点. 团簇的理论研究有助于揭示物质从微观的单个原子和分子到宏观凝聚态的演变规律, 为微观尺度材料的设计和改性提供理论支持[1]. 硼是缺电子元素的一个例子, 其原子核外有3个价电子, 因此显示出多种化学结构和键合模式. 硼的缺电子特性导致平面硼簇倾向于形成多中心的二电子键, 不同的连接方式导致多种团簇结构的形成[2–4]. 实验和理论研究均表明,
$ {\text{B}}_n^ - $ (n = 2—38, 41, 42) 具有准平面或平面结构[5–8], 而中性的Bn具有准平面、平面、轮状、管状或其他结构[2,3,8,9], 21世纪初, Boustani等[2,10–12]对中小尺寸纯硼阴离子团簇($ {\text{B}}_n^ - $ , n = 3—42)进行光电子能谱(PES)实验和理论计算. 基于对纯硼团簇的研究, Islas等[13,14]进一步将单一或多种元素掺入硼基团簇中, 该策略已被成功证实可以有效调节纯硼团簇的化学成键与几何结构, 从而产生一系列具有独特性能的硼基掺杂团簇. Saha等[15]研究发现, 单一过渡金属Co, Rh, Ir掺杂到$ {\text{B}}_{{12}}^ - $ 团簇中会形成独特的三维 “伞状” 结构. 2020年, 李世雄等[16]基于粒子群优化算法程序CALYPSO研究了掺Be硼团簇$ {\text{BeB}}_n^{{\text{0/}} - } $ (n = 10—15)的基态结构与性质, 发现了具有较强的非线性光学响应掺杂团簇$ {\text{BeB}}_{{13}}^ - $ 和$ {\text{BeB}}_{{14}}^ - $ .在各种掺杂元素中, 稀土元素因其改变无机纳米材料的晶相、形貌、尺寸和电子结构的能力而受到广泛关注[17]. 稀土元素具有独特的4f电子层结构, 这是稀土掺杂团簇具有广泛的光学、电学、磁学和催化性能的重要原因[18], 团簇组成的掺杂纳米材料在许多技术应用中具有发展前景[19–24]. 在过去的十年中, Li等[25–31]对稀土元素掺杂的硼基团簇进行了大量的实验和理论研究. Li等[32]利用密度泛函理论, 结合光电子能谱实验和理论计算, 研究了一系列镧系元素掺杂的硼团簇 (
$ {\text{REB}}_{8}^ - $ , RE = La, Pr, Tb, Tm, Yb) 的最低能量异构体的几何结构, 电子性质和成键方式. 他们发现了$ {\text{LnB}}_{8}^ - $ 和$ {\text{PrB}}_{8}^ - $ 共存的两种类型结构, 为设计具有可调节性和磁性的镧系硼化物提供了有效依据. 因此, 硼团簇的稀土元素掺杂是多样化硼团簇结构形式并拓展其性能的有效方法. 在此基础上, 本文采用人工蜂群算法结合密度泛函理论, 在PBE0/RE/SDD//B/6-311+G*水平下系统研究了稀土掺杂阴性硼团簇$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)的几何结构、稳定性、电子性质和芳香性. -
本文采用人工蜂群算法(artiffcial bees colony, ABC)[33]结合密度泛函理论(density functional theory, DFT), 在Gaussian 09程序下[34]完成对稀土掺杂硼阴性团簇
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)的全局优化, 通过多功能波函数分析程序Multiwfn[35]进行性质分析, 并用可视化分子动力学软件(visual molecular dynamics, VMD)[36]进行可视化处理. 本文对于稀土掺杂硼团簇$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)的计算采用以下3个步骤.1) 初始结构的预优化 对La原子和Sc原子分别采用ECP46MWB[37]和ECP10MDF基组[38], 对B原子采用3-21G基组[39]进行全局优化, 进行平面结构与立体结构各800次的随机搜寻. 计算选择PBE0交换关联泛函[40], 简记为PBE0/ La/ECP46MWB//B/3-21G和PBE0/Sc/ECP10MDF//B/3-21G.
2) 结构的二次优化 将一次优化得到的所有异构体按总能量的高低, 由低到高进行排序. 在此排序的基础上, 对每种尺寸下的体系选择能量最低的20个不同的异构体结构, 进行二次优化, 选择的基组与计算水平如下: 对掺杂原子均采用包含相对论效应的Stuttgart-Dresden大核赝势基组SDD[41]; 对B原子采用6-311+G*基组[42], 计算方法可写为PBE0/RE/SDD//B/6-311+G*.
3) 最优结构的确定 按照二次优化[PBE0/RE/SDD//B/6-311+G*]的结果, 将总能量从低到高的方式进行再次排序, 并计算它们相对能量的大小, 将相对能量最低的4个的异构体认为是可能的潜在的候选结构. 计算中考虑了自旋多重度对团簇能量的影响, 其中
$ {\text{ScB}}_{8}^ - $ 体系在自旋多重度为3时最稳定, 为开壳层结构, 其余体系均在自旋多重度为1时最为稳定, 为闭壳层结构. -
图1为稀土掺杂硼团簇
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)在PBE0/RE/SDD//B/6-311+G*计算水平下, 能量最低结构和其他低能异构体, 相对能量以及点群对称性. 图中所列的结构依据相对能量由低到高的顺序进行排序, 在分子式后用Ⅰ, Ⅱ, Ⅲ, Ⅳ表示. 由图1可见, 团簇$ {\text{LaB}}_{6}^ - $ 全局能量最低团簇$ {\text{LaB}}_{6}^ - $ -Ⅰ具有C2对称性, 团簇呈 “船形” 结构, 掺杂的La原子位于“船”的顶部中心. 异构体$ {\text{LaB}}_{6}^ - $ -Ⅱ和$ {\text{LaB}}_{6}^ - $ -Ⅲ由于它们平面和准平面的结构而具有更高的对称性(Cs和C2V).$ {\text{LaB}}_{6}^ - $ -Ⅱ可以看作是平面结构$ {\text{LaB}}_{6}^ - $ -Ⅲ中的La原子向B原子所在平面上方移动一定的角度得到的异构体, 而由于较高的对称性,$ {\text{LaB}}_{6}^ - $ -Ⅱ和$ {\text{LaB}}_{6}^ - $ -Ⅲ的能量比能量最低结构分别高0.06 eV和0.19 eV. 锥形异构体$ {\text{LaB}}_{6}^ - $ -Ⅳ表现出C2V对称性, La原子位于棱锥的顶点位置, 其余B原子位于底面. 中间4个B原子呈矩形排列, 长边B—B键长为2.01 Å, 短边B—B键长为1.61 Å. 另外两个B原子对称分布在两长边外侧, 与矩形的长边构成对称的等腰三角形, 腰的键长为1.55 Å. 同时, 异构体能量比全局最小结构高0.86 eV. 对于$ {\text{LaB}}_{6}^ - $ 团簇体系, 其4个低能异构体的结构与$ {\text{LaB}}_{6}^ - $ 团簇一致, 可以看作是$ {\text{LaB}}_{6}^ - $ 团簇中La原子被Sc原子取代得到, 相对能与对称性见图1.$ {\text{LaB}}_{8}^ - $ 团簇体系的基态结构如图中$ {\text{LaB}}_{8}^ - $ -Ⅰ所示, 展现出了Cs对称性, 异构体表现出“筝形”的结构特征, 6个B原子组成六边形, 其余一个B原子位于六边形中心, 一个B原子位于六边形外侧. 而$ {\text{LaB}}_{8}^ - $ -Ⅱ展现出了更高的对称性(C7V), 其结构可看作是 “伞状” 的立体结构, 底部外围的7个B原子组成了正七边形的7个顶点, 它们之间的距离(B—B键键长) 为1.54 Å, 另一个B原子位于七边形的中心, La原子位于 “伞状” 结构的 “伞柄” 处. 另外两个异构体$ {\text{LaB}}_{8}^ - $ -Ⅲ(Cs) 和$ {\text{LaB}}_{8}^ - $ -Ⅳ(C1) 显示出了较为复杂的构型, 并可推断出它们不太可能被实验检测到. 因为它们的相对能量分别达到1.40 eV和1.52 eV.$ {\text{ScB}}_{8}^ - $ 团簇相较于$ {\text{LaB}}_{8}^ - $ 团簇呈现出能级反转现象.$ {\text{ScB}}_{8}^ - $ -Ⅲ具有C8V对称性, 其结构为一个正八边形, 由8个B原子组成顶点, B—B键长为1.55 Å, Sc原子位于正八边形中心上方, Sc—B键长为2.40 Å. 类似于$ {\text{LaB}}_{8}^ - $ -Ⅲ, 异构体$ {\text{ScB}}_{8}^ - $ -Ⅳ(Cs)展示出复杂的构型, 能量比基态高出1.54 eV, 推测不太可能在实验中检测到.通过以上对
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)结构特征的分析, 表明了低能异构体中既存在平面结构也存在三维立体构型, 且掺杂原子总是倾向于占据$ {\text{REB}}_n^ - $ 团簇的表面位置. 在本文的计算中并未发现掺杂原子位于硼基团簇内部的内嵌式低能异构体. 各团簇低能异构体的笛卡尔坐标见补充材料表S1—S4 (online ). -
定域化轨道指示函数(localized orbital locator, LOL)可用于反映电子在团簇不同区域中的定位函数. 图2为
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)低能异构体的LOL函数等值面图. 从三维立体图像可见, 16种不同结构的绿色的等值面均占据在B原子之间, 意味着电子倾向于局域在B—B之间的区域中, 表明了B—B之间以共享电子对的方式进行相互作用, 这意味着B—B之间很有可能以共价键的形式连接. 有趣的是, 还可以观察到每个Sc原子都被绿色等值面包围着, 这表明Sc原子周围有较大空间范围的电子分布, La原子周围则没有孤对电子聚集的现象. 除此之外, RE—B之间的相互作用在这张图中是无法判断的, 因为在RE原子和B原子之间电子定域化程度较低, 这说明RE—B之间的电子定域性不如B—B之间的电子定域性. -
基于广义库普曼定理[43], 本文研究在PBE0/RE/SDD//B/6-311+G*计算水平下异构体的模拟光电子能谱, 并同时计算了这些低能异构体的第一垂直拆分能(VDE). VDE被定义为中性结构 (保留阴性团簇优化后的构型) 的能量与阴性结构能量的差值. 倘若光电子能谱的VDE, 谱峰数量及其相对位置能较好地契合这一标准, 便可将其视作在实验中能够稳定存在的异构体. 图3为
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)团簇低能异构体的模拟光电子能谱, 与Li等[32]实验测出$ {\text{LaB}}_{8}^ - $ 的光电子能谱同时绘制在图3中以便于比较, 然而, 其余团簇体系的实验光电子能谱暂未见报道, 实验光电子能谱中的字母X代表第一个实验VDE值. 各体系理论VDE值详细结果见补充材料表 S5(online ). 由于计算中不考虑非谐振和非绝热相互作用, 计算得到的峰值强度无法与实验数据进行定量比较, 因此不考虑峰值强度, 仅考虑光谱峰值的相对位置.对于
$ {\text{LaB}}_{8}^ - $ 团簇, 结合了理论与实验光电子能谱进行比较, 最低能量异构体$ {\text{LaB}}_{8}^ - $ -Ⅰ, 其理论光电子能谱呈现出6个峰值, 位于2.29—5.23 eV之间. 计算出的VDE值为2.29 eV, 这与实验光电子能谱的结果基本保持一致(2.40 eV), 经过很窄的一段能隙后, 在2.61 eV处又出现了能量强度相似的谱峰, 这与实验谱中的2.77 eV也保持了一致. 对于剩余谱峰, 具体来说, 谱峰位置分别为3.08 eV, 4.20 eV, 4.73 eV和5.25 eV, 非常接近实验中所观测到的峰值(3.18 eV, 4.11 eV, 4.70 eV和5.11 eV). 理论峰值与实验峰值得到了良好的匹配, 此外, 对于$ {\text{LaB}}_{8}^ - $ 体系的另外三个低能异构体, 都不具备与实验谱相似的光谱模式, 通过仔细比较异构体$ {\text{LaB}}_{8}^ - $ -Ⅱ,$ {\text{LaB}}_{8}^ - $ -Ⅲ和$ {\text{LaB}}_{8}^ - $ -Ⅳ对实验光电子能谱的贡献, 发现可以忽略不计, 同时, 它们的相对能均大于0.40 eV, 因此,$ {\text{LaB}}_{8}^ - $ -Ⅰ被认为是$ {\text{LaB}}_{8}^ - $ 体系的全局最优结构.对于
$ {\text{LaB}}_{6}^ - $ 团簇, 模拟得到的光电子能谱与$ {\text{LaB}}_{8}^ - $ 团簇表现出类似的光谱特征, 均呈现出6个明显的谱峰, 谱峰值位于1.74—4.73 eV之间.$ {\text{ScB}}_{6}^ - $ 团簇与$ {\text{LaB}}_{6}^ - $ 表现出类似的光谱特征. 模拟得到的VDE为2.08 eV. 对于$ {\text{ScB}}_{8}^ - $ 团簇, 模拟得到的光电子能谱与$ {\text{LaB}}_{8}^ - $ 团簇表现出的光谱特征有很大的差别, 这是由于结构的巨大差异导致电子构型的巨大差异. 其中第1个谱峰对应的VDE为1.91 eV.简言之, 通过对
$ {\text{LaB}}_{8}^ - $ 体系的理论谱与实验谱进行比较, 发现能量最低的结构在团簇模拟的光电子能谱与实验数据中有良好的一致性, 计算得到的VDE接近实验值.$ {\text{LaB}}_{8}^ - $ -Ⅰ被确认为该体系的全局最优结构, 这也验证了选择PBE0/RE/SDD//B/6-311+G*泛函基组的正确性, 并且此基组适用于其他团簇体系. 通过对上述模拟光电子能谱和数据进行详尽分析, 可推断所研究的能量最低异构体是掺杂团簇的全局最优结构. 同时, 期待本研究获得的模拟光电子能谱数据与理论结果能为未来实验提供可靠的理论基础. -
图4为闭壳层结构
$ {\text{LaB}}_{6}^ - $ 的芳香性分析(AdNDP)分析图, 对全局优化后能量最低结构$ {\text{LaB}}_{6}^ - $ -Ⅰ用AdNDP方法进行化学键分析, 揭示了其结构外围的4个2c-2e B—B σ键, 它们的轨道占据数在1.85—1.90 |e|之间, 和两个3c-2e La—B σ键, 它们的轨道占据数为1.94 |e|. 此外, 还有3个7c-2e π键和3个7c-2e σ键, 他们的轨道占据数均为2.00 |e|. 对于全局的7c-2 e轨道, 3个全局π键和3个全局σ键构成了6个π电子和6个σ电子, 因此可以认为它是σ-π双芳香簇, 结构呈现出芳香性特征, 这也是它能够作为全局优化后能量最低结构的原因. 对于$ {\text{LaB}}_{6}^ - $ -Ⅳ, 其外围有3对对称的2c-2e B—B σ键, 它们的轨道占据数在1.90—1.95 |e|之间. 在全局轨道上, 有2个7c-2e π键, 以及3个7c-2e σ键, 与次低能异构体类似, 2个全局π键和3个全局σ键分别构成了4个π电子和6个σ电子, 当体系的π电子数为4n+2, 而体系的σ电子数为4n时, 相应的电子稳定性会降低, 由此形成的化合物具有冲突芳香性. 它是σ型芳香簇而π型反芳香簇, 具有冲突芳香性, 这可以作为解释其全局能量高于前3种异构体的依据. 不难发现$ {\text{LaB}}_{6}^ - $ -Ⅳ与$ {\text{LaB}}_{6}^ - $ -Ⅰ在几何结构上存在一定的相似性, 两者的形状都来源于$ {\text{B}}_{6}^ - $ 的能量最低异构体[44]. 不同之处在于$ {\text{LaB}}_{6}^ - $ -Ⅳ保留了$ {\text{B}}_{6}^ - $ 的平面性, 而由于$ {\text{LaB}}_{6}^ - $ -Ⅰ中La与两端B原子的距离, 使得电子在此区域内更倾向于形成定域性更强的3c-2e σ键而不是全局离域σ键. 另外, 经过尝试发现, 由于存在两个外围键距极短的B—B键, 其电子定域性不强, 无法划分为2c-2e σ键, 而倾向于构成全局离域键.反观次低能异构体
$ {\text{LaB}}_{6}^ - $ -Ⅱ, 其外围有6个两两对称的2c-2e B—B σ键, 它们的轨道占据数在1.92—1.95 |e|之间, 和1个5c-2e π键, 它们的轨道占据数为2.00 |e|. 此外, 还有1个7c-2e π键, 以及3个7c-2e σ键, 他们的轨道占据数均为2.00 |e|. 对于全局的7c-2 e轨道, 1个全局π键和3个全局σ键分别构成了2个π电子和6个σ电子, 它既属于σ型芳香簇, 又具有 π 型反芳香簇的性质, 其结构总体上依旧展现出芳香性特征. 这一特性能够很好地解释它的能量仅比全局最低结构略高 0.06 eV 的现象.对于平面结构的
$ {\text{LaB}}_{6}^ - $ -Ⅲ, 其外围有3对对称的2c-2e B-B σ键, 它们的轨道占据数在1.92—1.95 |e|之间. 在全局轨道上, 有2个7c-2e π键, 以及3个7c-2e σ键, 与次低能异构体类似, 2个全局π键和3个全局σ键分别构成了4个π电子和6个σ电子, 它是σ型芳香簇而π型反芳香簇, 具有冲突芳香性, 相对能量值为0.19 eV.图5为同是闭壳层结构的
$ {\text{ScB}}_{6}^ - $ -Ⅰ, 价电子数为22, 对全局优化后能量最低结构$ {\text{ScB}}_{6}^ - $ -Ⅰ用AdNDP方法进行化学键分析, 揭示了其结构外围的4个2c-2e B—B σ键, 它们的轨道占据数在1.85—1.89 |e|之间, 和两个关于中心Sc原子对称的3c-2e La—B σ键, 它们的轨道占据数为1.96 |e|. 此外, 还有3个7c-2e π键和3个7c-2e σ键, 他们的轨道占据数均为2.00 |e|. 对于全局的7c-2e轨道, 3个全局π键和3个全局σ键构成了6个π电子和6个σ电子, 它们均符合4n+2休克尔规则, 因此可以认为其是σ-π双芳香簇, 结构呈现出芳香性特征, 这也是它能够作为全局优化后能量最低结构的原因.图6为相对能量0.05 eV的
$ {\text{ScB}}_{6}^ - $ -Ⅲ, 其外围有6个2c-2e B—B σ键, 它们的轨道占据数在1.92—1.95 |e|之间, 在全局轨道上, 有2个7c-2e π键, 以及3个7c-2e σ键, 他们的轨道占据数均为2.00 |e|. 对于全局的7c-2e轨道, 2个全局π键和3个全局σ键分别构成了4个π电子和6个σ电子, 6个σ电子符合4n+2休克尔规则, 而4个π电子并不符合休克尔规则, 它是σ型芳香簇而π型反芳香簇. -
为了更好地理解RE原子的掺杂对团簇分子轨道的影响, 将
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)全局最优结构的总态密度图(total density of states, TDOS)绘制在图7中, 同时还绘制了RE原子和B6/B8框架的局部态密度图(partial density of states, PDOS), 从而评估它们对TDOS的贡献, 图中的竖直虚线表示团簇前沿分子轨道(HOMO和LUMO轨道) 的位置情况. 由图7可知, 对于4个体系而言, 掺杂原子的PDOS曲线与B6/B8的PDOS曲线相比通常较低, 这表明分子轨道的组成主要由B6/B8框架贡献. 掺杂原子RE几乎不参与分子轨道的形成. 这一现象对于闭壳层体系$ {\text{LaB}}_{8}^ - $ 尤为显著. 但也有例外的是开壳层结构的$ {\text{ScB}}_{8}^ - $ , 掺杂原子Sc在α-HOMO和β-LUMO轨道的组成中异常活跃, 并对团簇的能量较低分子轨道有显著贡献. 同时, α轨道与β轨道的TDOS在能量较高的区域有明显的差异, 说明该体系有一定的自旋极化现象, 是潜在的磁性团簇基元.团簇的HOMO-LUMO能隙反映了电子从占据轨道跃迁到未占据轨道所需要的能量. 对于闭壳层的
$ {\text{LaB}}_{6}^ - $ ,$ {\text{LaB}}_{8}^ - $ 和$ {\text{ScB}}_{6}^ - $ , 它们的HOMO-LUMO能隙分别为2.02 eV, 2.27 eV和1.67 eV, 这在一定程度上表明其参与化学反应的能力较强, 对于开壳层的$ {\text{ScB}}_{8}^ - $ , 存在较大的HOMO-LUMO能隙, α轨道与β轨道的HOMO-LUMO能隙分别为2.26 eV和4.08 eV, 这可以作为团簇化学稳定性较强的依据. -
利用人工蜂群算法结合第一性原理计算, 研究了稀土掺杂阴离子硼团簇
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)的稳定结构, 得到了能量最低的基态结构, 主要研究结果如下.1)
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6)的基态结构均具有C2对称性, 掺杂的RE原子位于顶部中心的 “船形” 结构. 通过与实验光电子能谱的比较, 证实$ {\text{LaB}}_{8}^ - $ 的基态结构呈现类似“筝形” 的三维结构, 而$ {\text{ScB}}_{8}^ - $ 的基态结构是由Sc原子位于 “伞柄” 处而形成的具有C7V点群对称的 “伞状” 结构.2) LOL分析表明, B—B之间是以共价相互作用为主导的成键模式, 而RE—B之间的电子定域性不如B—B之间的电子定域性.
3) AdNDP分析表明,
$ {\text{LaB}}_{6}^ - $ -Ⅰ和$ {\text{ScB}}_{6}^ - $ -Ⅰ是π电子和σ电子均符合休克尔(4n+2)规则的σ-π双芳香簇, 结构呈现出芳香性特征, 这也是它们能够作为全局优化后能量最低结构的主因. 而$ {\text{LaB}}_{6}^ - $ 和$ {\text{ScB}}_{6}^ - $ 的其余低能异构体则普遍具有反芳香性, 这也解释了它们的相对能量较高的现象.4)态密度分析表明,
$ {\text{ScB}}_{8}^ - $ 态密度谱呈现出自旋极化现象, 表明其有望成为磁性纳米材料的组装基元.综上所述, 本文的理论预测极大程度地丰富了稀土掺杂硼基团簇的结构, 进一步加深了对稀土掺杂硼基团簇性质与演变规律的认识, 为未来实验奠定了理论基础.
稀土掺杂硼团簇$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) 的几何及电子结构
Geometry and electronic structures of rare earth-doped boron-based clusters $ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)
-
摘要: 稀土掺杂硼团簇因其特殊的光学, 电学和磁学性质受到广泛关注. 本文采用人工蜂群算法结合密度泛函理论, 在PBE0/RE/SDD//B/6-311+G*水平下研究了稀土掺杂阴离子硼团簇$ {\text{REB}}_n^ - $(RE = La, Sc; n = 6, 8) 的几何结构、电子性质、稳定性和芳香性. 计算结果表明, 阴离子$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6) 的基态结构具有C2对称性, 掺杂的镧系原子位于顶部中心形成“船形”结构. 通过与实验光电子能谱的比较, 证实$ {\text{LaB}}_{8}^ - $ 的基态结构类似于三维的“筝形”结构, 而$ {\text{ScB}}_{8}^ - $ 的基态结构则是Sc原子位于“伞柄”处形成的具有C7V对称性的“伞状”结构. B—B之间存在通过共享电子对的相互作用, 而RE—B之间的电子定域性不如B—B之间的电子定域性. 模拟得出的光电子能谱峰值位置与实验结果的吻合度较高, 充分证实了研究获取的全局能量最低结构与实验观测结构的一致性. $ {\text{LaB}}_{6}^ - $ 和$ {\text{ScB}}_{6}^ - $ 的最低能量结构均为σ-π双芳香簇, 表现出明显的芳香性. 此外, 分别计算了$ {\text{REB}}_n^ - $(RE = La, Sc; n = 6, 8)的总态密度, 以及RE原子和硼簇的局部态密度, 并对其轨道能级密度进行了评估. 开壳层的$ {\text{ScB}}_{8}^ - $态密度谱呈现出自旋极化现象, 这表明其作为基元可以组装成具有磁性的纳米材料. 这些对稀土掺杂硼团簇的研究有助于深入理解纳米材料的结构和性质演变规律, 为设计具有实际价值的纳米材料提供了重要的理论支持.Abstract: Rare earth doped boron clusters have attracted much attention due to their special optical, electrical and magnetic properties. The geometric structures, stability, electronic properties and aromaticity of negative rare earth doped boron clusters $ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) are investigated with the artificial bee colony algorithm combined with density functional theory calculations at the PBE0/RE/SDD//B/6-311+G* level of theory. Calculations show that the ground state structures of $ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) are all of C2 symmetry, and the doped lanthanide atom is located in a “boat-shaped” structure at the top center. By comparing with the experimental photoelectron spectra, it is confirmed that the ground state structure of $ {\text{LaB}}_{8}^ - $ is a “zither-like” three-dimensional structure, and the ground state structure of $ {\text{ScB}}_{8}^ - $ is an “umbrella” structure with C7V symmetry formed by the scandium atom at the “umbrella handle”. The electron localization between RE—B is not as good as that between B—B. The simulated photoelectron spectra have similar spectral characteristics to the experimental results. The lowest energy structures of $ {\text{LaB}}_{6}^ - $ and $ {\text{ScB}}_{6}^ - $ are σ-π double aromatic clusters, and the structures exhibit aromaticity. The density of states of low-energy isomers shows that the open shell $ {\text{ScB}}_{8}^ - $ density of states spectrum exhibits spin polarization phenomenon, which is expected to assemble magnetic material components. These studies contribute to understanding the evolution of structure and properties of nanomaterials, and provide important theoretical support for designing nanomaterials with practical value.
-
Key words:
- density functional theory /
- rare earth doped clusters /
- structural optimization /
- aromaticity .
-

-
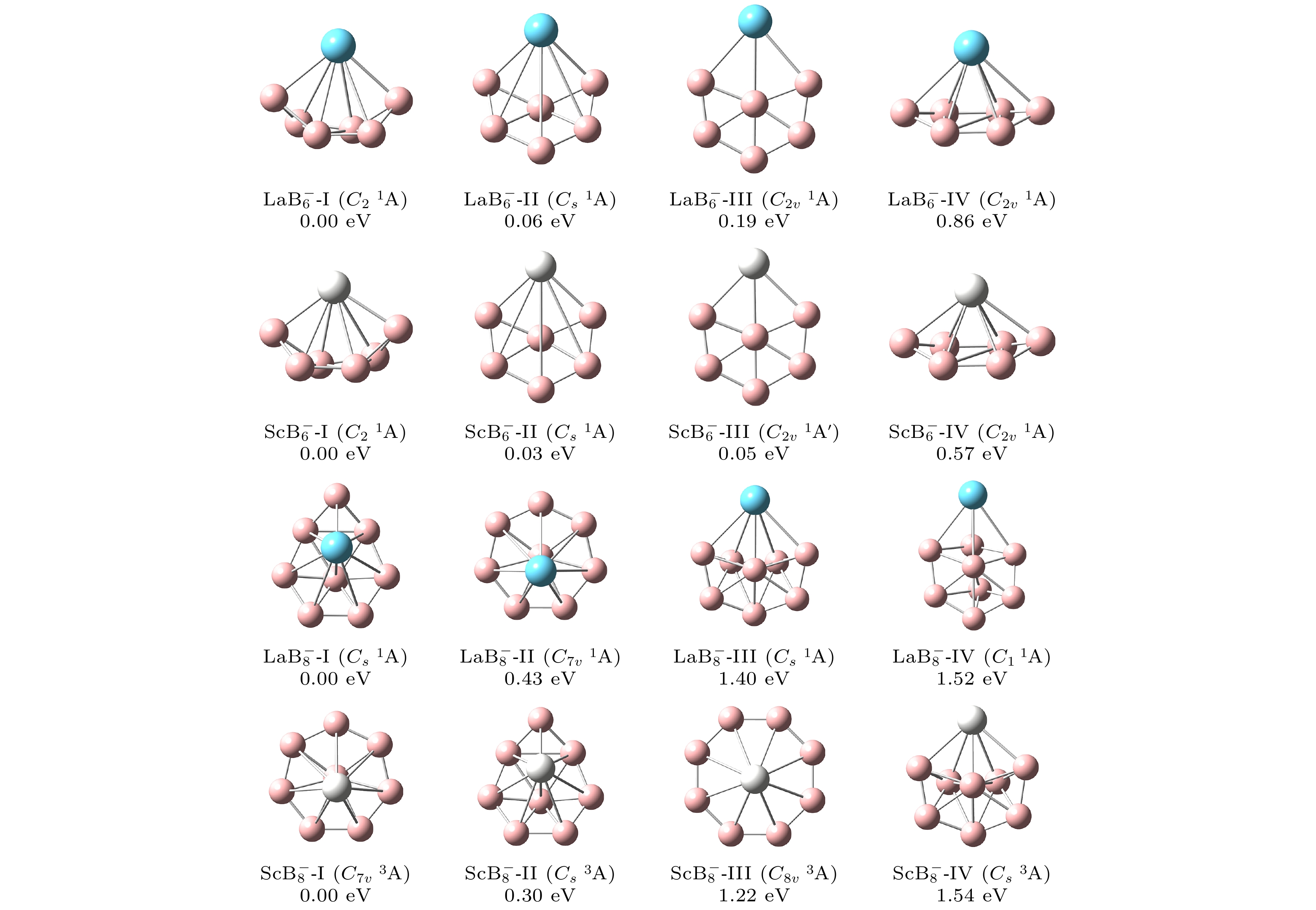
图 1 在PBE0/RE/SDD//B/6-311+G*理论水平下,
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) 团簇低能异构体的结构、点群对称性、相对能Figure 1. Structures, symmetry point group and relative energy (eV) of the lower-lying isomers for
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) clusters at the PBE0/RE/SDD//B/6-311+G* level of theory.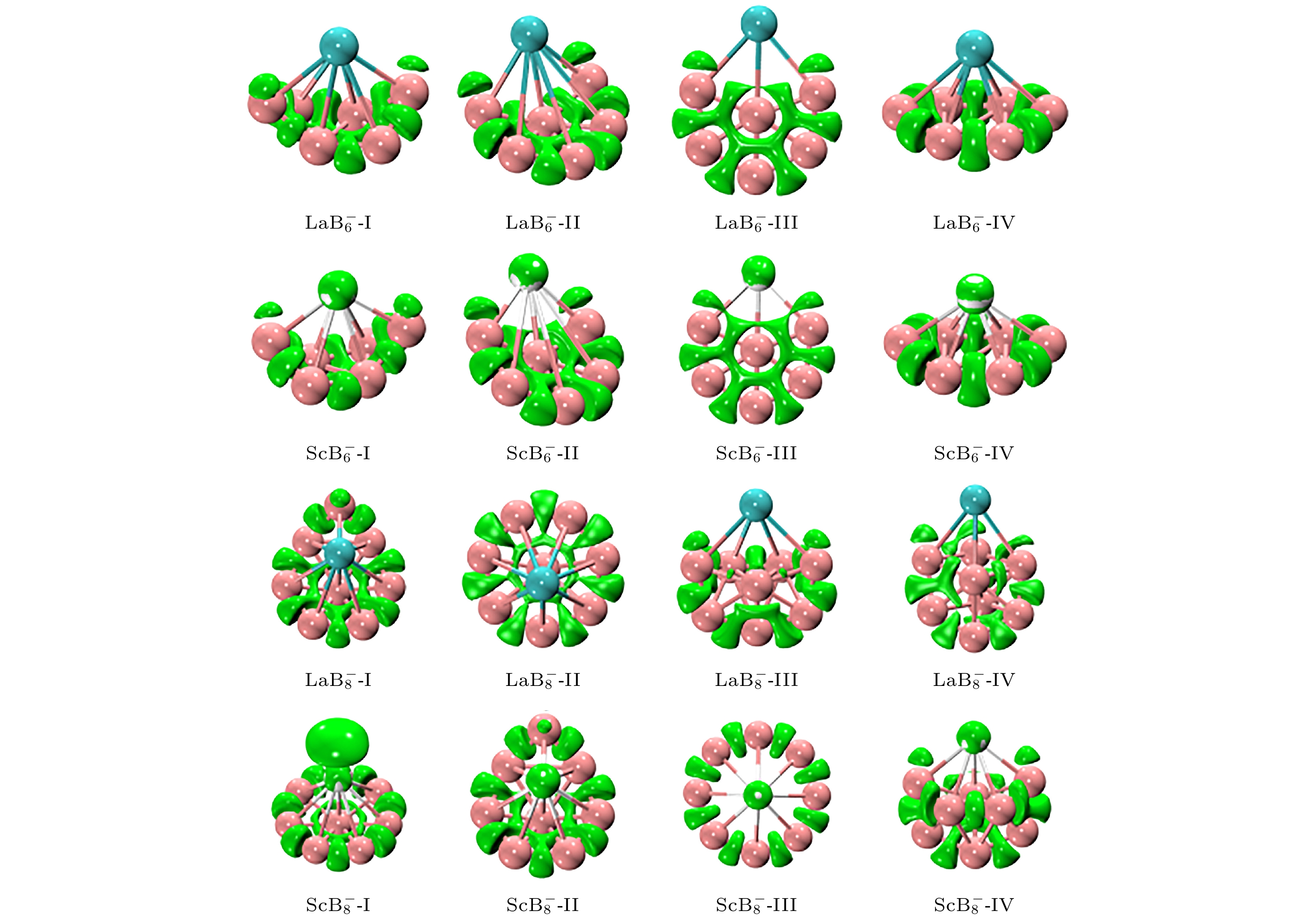
图 2
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)团簇低能异构体的定域化轨道指示函数等值面图Figure 2.
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) isosurface of localized orbital locator function of low energy isomers.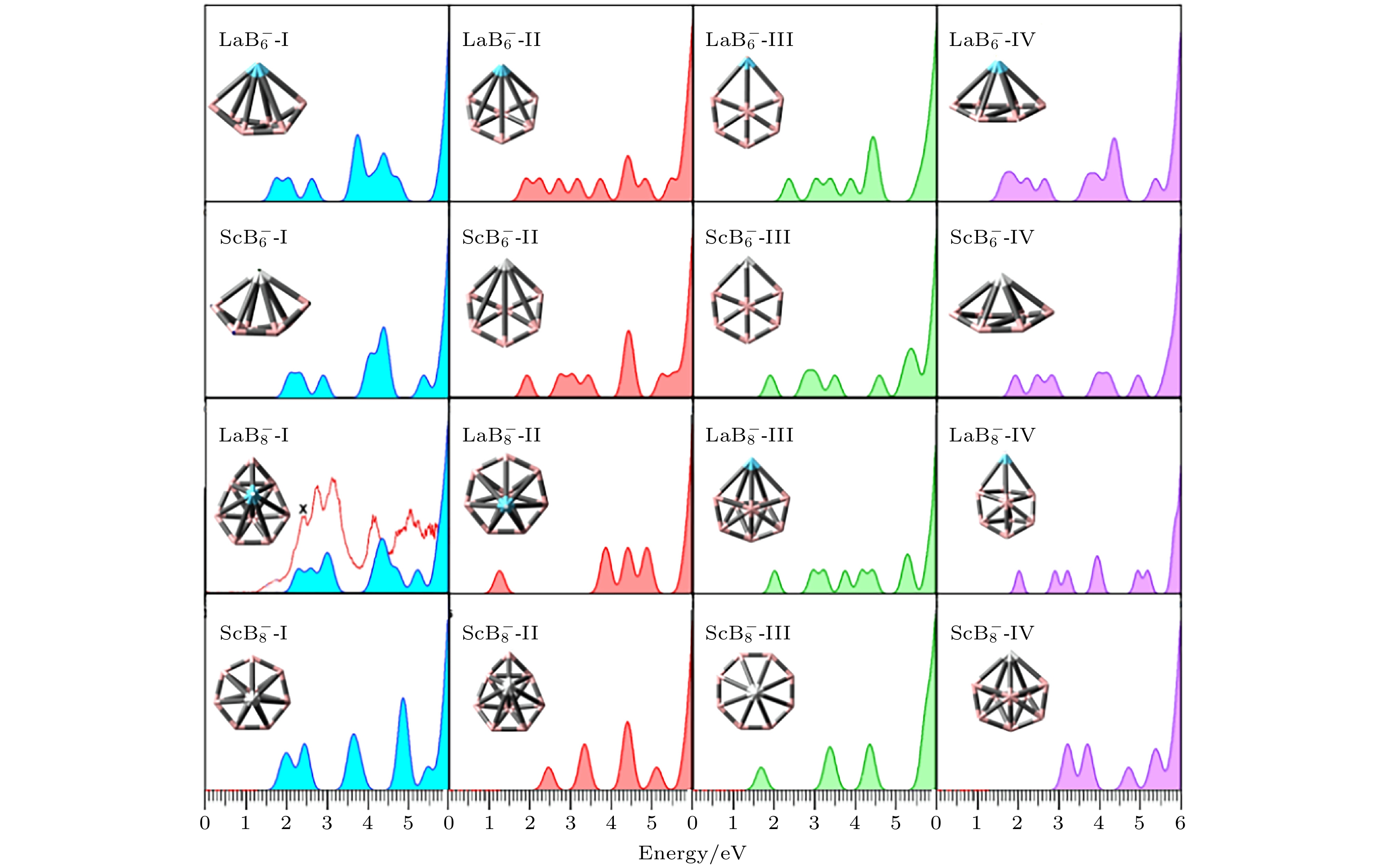
图 3
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8)团簇低能异构体的模拟光电子能谱图,$ {\text{LaB}}_{8}^ - $ 的实验光电子能谱来自于文献[32], 共同绘制在$ {\text{LaB}}_{8}^ - $ -Ⅰ处用红色曲线表示Figure 3. Simulated PES spectra for low-lying isomers of
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) clusters, the experimental PES spectra of the anionic ground state structure of$ {\text{LaB}}_{8}^ - $ was obtained from Ref. [32].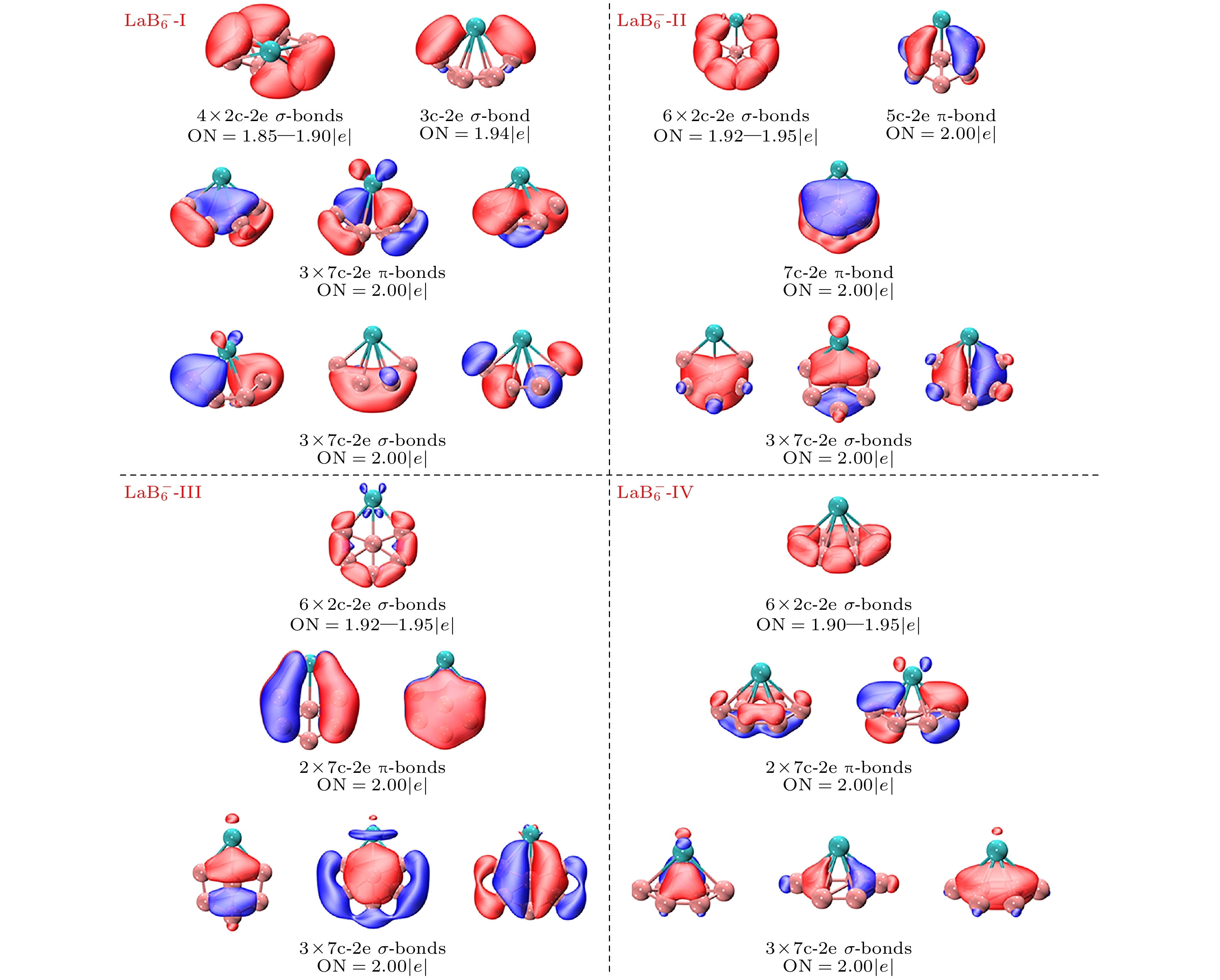
图 4
$ {\text{LaB}}_{6}^ - $ 的芳香性分析图Figure 4. Adaptive natural density partitioning (AdNDP) bonding analyses of
$ {\text{LaB}}_{6}^ - $ .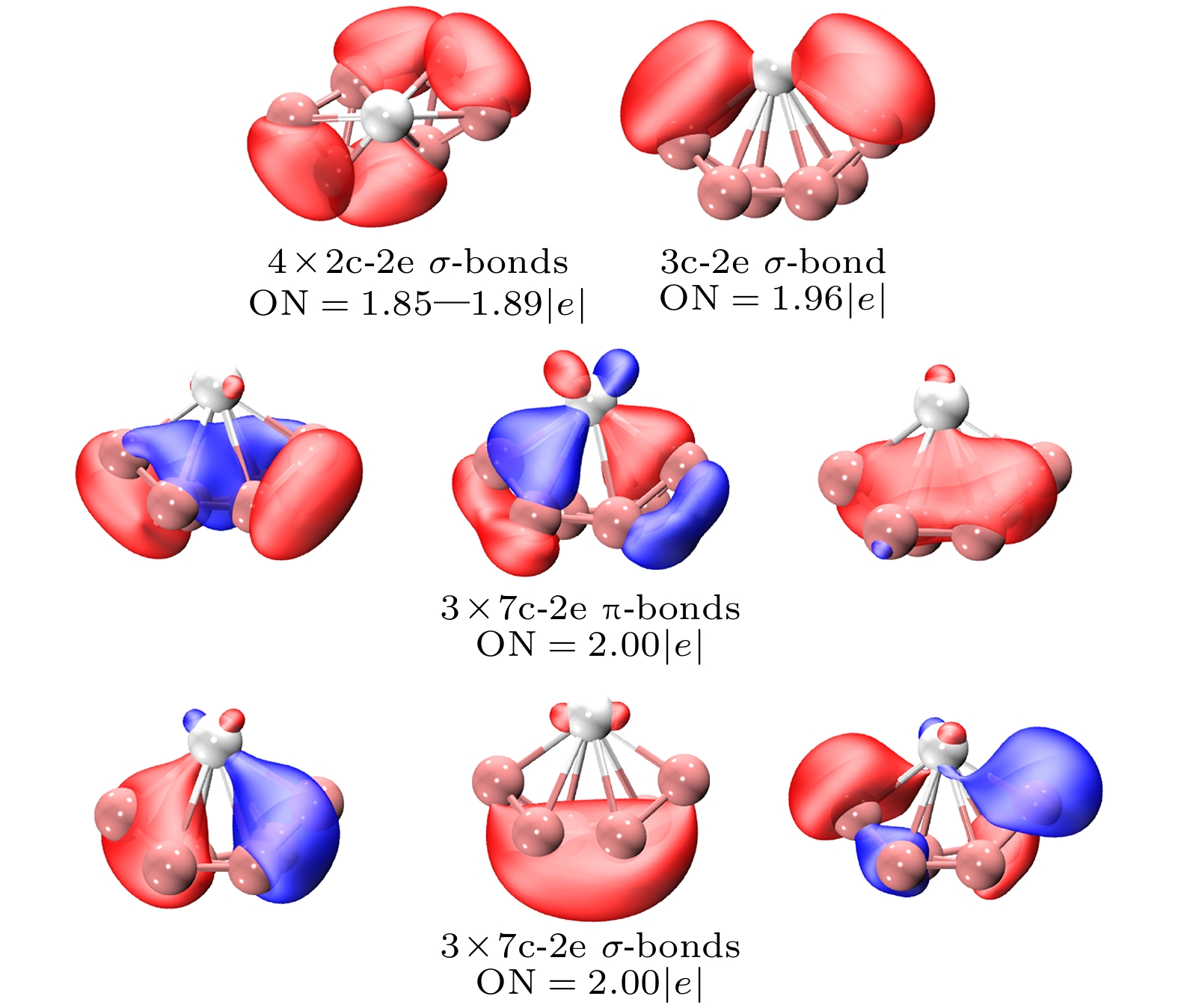
图 5
$ {\text{ScB}}_{6}^ - $ -Ⅰ的芳香性分析轨道图Figure 5. Adaptive natural density partitioning (AdNDP) bonding analyses of
$ {\text{ScB}}_{6}^ - $ -Ⅰ.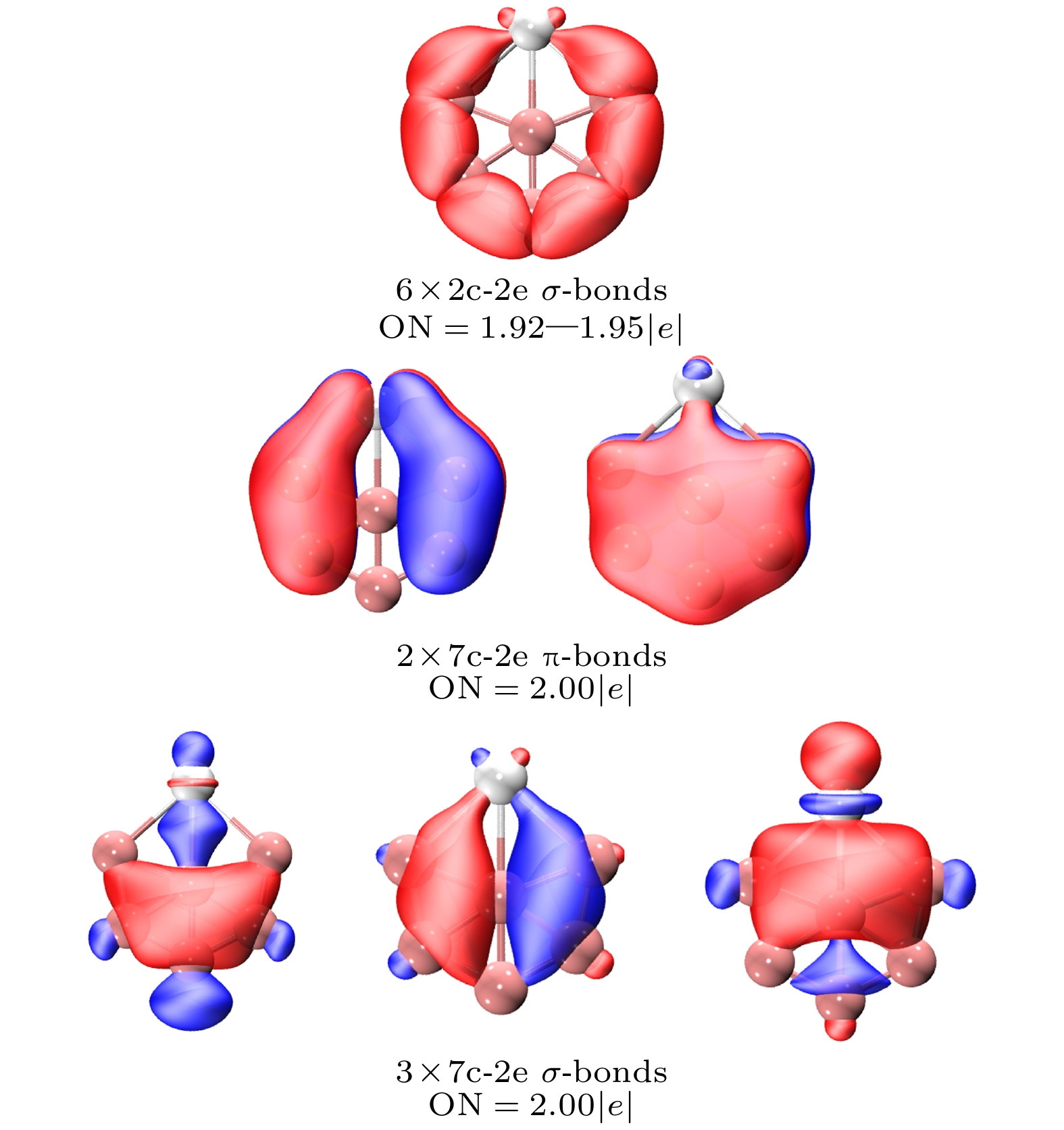
图 6
$ {\text{ScB}}_{6}^ - $ -Ⅲ的芳香性分析图Figure 6. Adaptive natural density partitioning (AdNDP) bonding analyses of
$ {\text{ScB}}_{6}^ - $ -Ⅲ.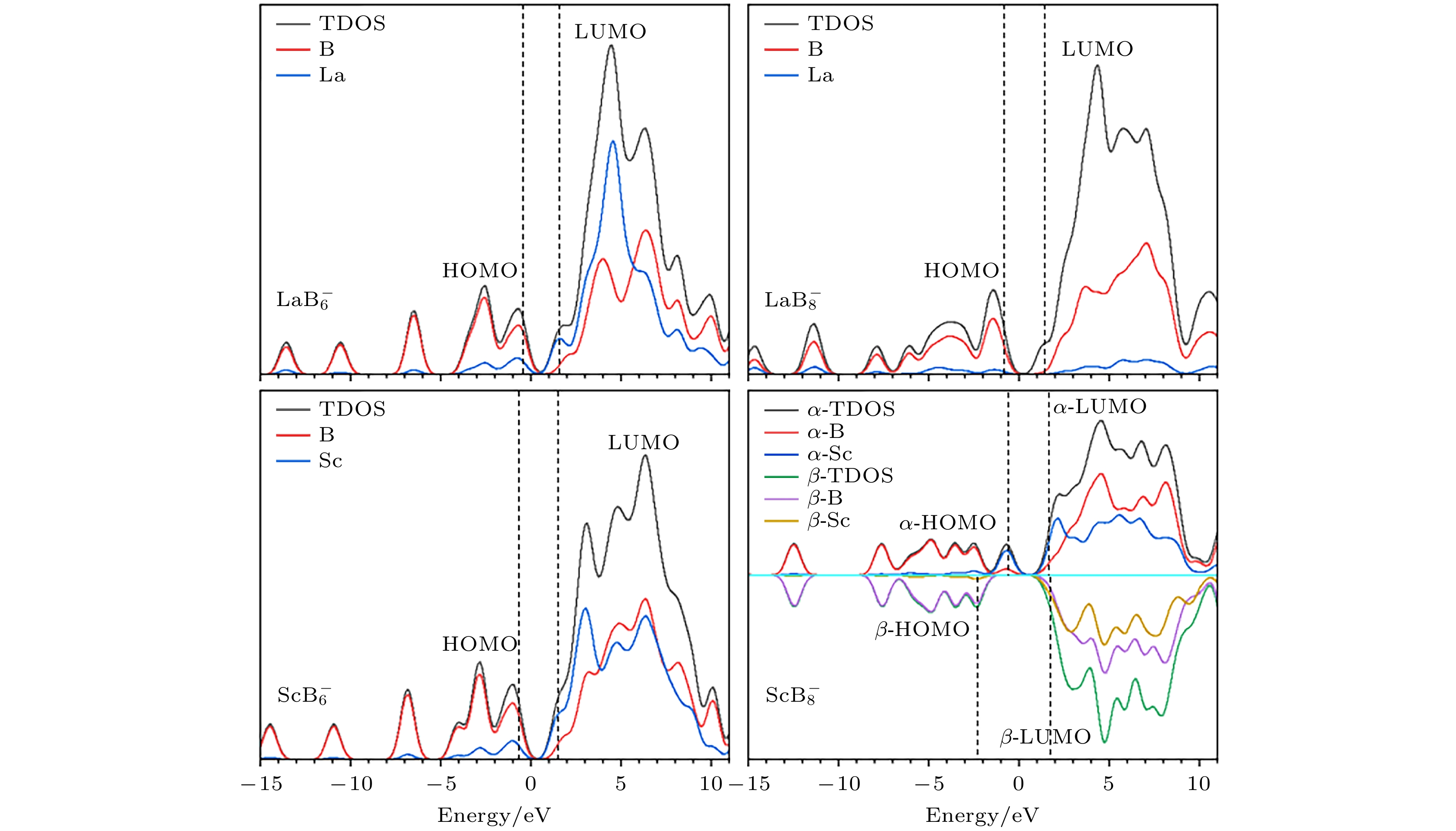
图 7 0.60 eV的半峰展宽下
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) 能量最低结构的总态密度图与局部态密度图Figure 7. Total density of states (TDOS) and partial density of states (PDOS) plot of the lowest energy structures of
$ {\text{REB}}_n^ - $ (RE = La, Sc; n = 6, 8) with a full width at half-maximum (FWHM) of 0.60 eV. -
[1] 张超江, 许洪光, 徐西玲, 郑卫军 2021 物理学报 70 023601 doi: 10.7498/aps.70.20201351 Zhang C J, Xu H G, Xu X L, Zheng W J 2021 Acta Phys. Sin. 70 023601 doi: 10.7498/aps.70.20201351 [2] Boustani I 1997 Phys. Rev. B 5 16426 [3] Kiran B, Bulusu S, Zhai H J, Yoo S, Zeng X C, Wang L S 2005 Proc. Natl. Acad. Sci. 102 961 doi: 10.1073/pnas.0408132102 [4] 李世雄, 张正平, 隆正文, 秦水介 2017 物理学报 6 103102 doi: 10.7498/aps.66.103102 Li S X, Zhang Z P, Long Z W, Qin S J 2017 Acta Phys. Sin. 6 103102 doi: 10.7498/aps.66.103102 [5] Sergeeva A P, Popov I A, Piazza Z A, Li W L, Romanescu C, Wang L S, Boldyrev A I 2014 Acc. Chem. Res. 47 1349 doi: 10.1021/ar400310g [6] Jian T, Chen X, Li S D, Boldyrev A I, Li J, Wang L S 2019 Chem. Soc. Rev. 48 3550 doi: 10.1039/C9CS00233B [7] Wang L S 2016 Int. Rev. Phys. Chem. 35 69 doi: 10.1080/0144235X.2016.1147816 [8] Bai H, Chen T T, Chen Q, Zhao X Y, Zhang Y Y, Chen W J, Li W L, Cheng L F, Bai B, Cavanagh J, Huang W, Li S D, Li J, Wang L S 2019 Nanoscale 11 23286 doi: 10.1039/C9NR09522E [9] 刘立仁, 雷雪玲, 陈杭, 祝恒江 2009 物理学报 58 5355 doi: 10.7498/aps.58.5355 Liu L R, Lei X L, Chen H, Zhu H J 2009 Acta Phys. Sin. 58 5355 doi: 10.7498/aps.58.5355 [10] Pham H T, Duong L V, Pham B Q, Nguyen M T 2013 Chem. Phys. Lett. 577 32 doi: 10.1016/j.cplett.2013.05.041 [11] Zhai H J, Alexandrova A N, Birch K A, Boldyrev A I, Wang L S 2003 Angew. Chem. Int. Ed. 42 6004 doi: 10.1002/anie.200351874 [12] Zhai H J, Zhao Y F, Li W L, Chen Q, Bai H, Hu H S, Piazza Z A, Tian W J, Lu H G, Wu Y B, Mu Y W, Wei G F, Liu Z P, Li J, Li S D, Wang L S 2014 Nature Chem. 6 727 doi: 10.1038/nchem.1999 [13] Islas R, Heine T, Ito K, Schleyer P V, Merino G 2007 J. Am. Chem. Soc. 129 14767 doi: 10.1021/ja074956m [14] Romanescu C, Galeev T R, Li W L, Boldyrev A I, Wang L S 2013 Acc. Chem. Res. 46 350 doi: 10.1021/ar300149a [15] Saha R, Kar S, Pan S, Martínez-Guajardo G, Merino G, Chattaraj P K 2017 J. Phys. Chem. A 121 2971 doi: 10.1021/acs.jpca.6b12232 [16] 李世雄, 陈德良, 张正平, 隆正文 2020 物理学报 69 193101 doi: 10.7498/aps.69.20200756 Li S X, Chen D L, Zhang Z P, Long Z W 2020 Acta Phys. Sin. 69 193101 doi: 10.7498/aps.69.20200756 [17] Li H F, Wang H Q, Zhang J M, Qin L X, Zheng H, Zhang Y H 2024 Molecules 29 1692 doi: 10.3390/molecules29081692 [18] 蒋贤明, 王怀谦, 曹宇, 孙之惠, 曹玉芳, 吴伟宾 2018 高等学校化学学报 39 1976 doi: 10.7503/cjcu20180218 Jiang X M, Wang H Q, Cao Y, Sun Z H, Cao Y F, Wu W B 2018 Chemical Journal of Chinese Universities 39 1976 doi: 10.7503/cjcu20180218 [19] Zheng H, Wang H Q, Li H F, Zhang J M, Zhang Y H, Qin L X, Mei X J, Jiang K L, Zeng J K, Zhang B, Wu W H 2024 Chem. Phys. 583 112321 doi: 10.1016/j.chemphys.2024.112321 [20] Wen S H, Zhou J J, Zheng K Z, Bednarkiewicz A, Liu X G, Jin D Y 2018 Nat. Commun. 9 2415 doi: 10.1038/s41467-018-04813-5 [21] Jiang L Y, Wang H Q, Li H F, Xie B, Zhang J M, Ji J Y 2023 Chem. Phys. 567 111819 doi: 10.1016/j.chemphys.2023.111819 [22] Wang H Q, Li H F 2014 RSC Adv. 4 29782 doi: 10.1039/C4RA03788J [23] Yi Z G, Luo Z C, Qin X, Chen Q S, Liu X G 2020 Acc. Chem. Res. 53 2692 doi: 10.1021/acs.accounts.0c00513 [24] Qin L X, Li H F, Xiao B W, Zhang J M, Zeng J K, Mei X J, Zhang Y H, Zheng H, Wang H Q 2023 Chem. Phys. 575 112064 doi: 10.1016/j.chemphys.2023.112064 [25] Li W L, Chen T T, Xing D H, Chen X, Li J, Wang L S 2018 Proc. Natl. Acad. Sci. 115 E6972 [26] Robinson P J, Zhang X X, McQueen T, Bowen K H, Alexandrova A N 2017 J. Phys. Chem. A 121 1849 doi: 10.1021/acs.jpca.7b00247 [27] Chen T T, Li W L, Li J, Wang L S 2019 Chem. Sci. 10 2534 doi: 10.1039/C8SC05443F [28] Zuo J N, Zhang L L, Chen B L, He K H, Dai W, Ding K W, Lu C 2024 J. Phys. Condens. Matter 36 015302 doi: 10.1088/1361-648X/acfc0c [29] Xiang Z Y, Luo Z J, Bi J, Jin S Y, Zhang Z Q, Lu C 2022 J. Phys. Condens. Matter 34 445302 doi: 10.1088/1361-648X/ac8b4f [30] Jin S Y, Sun W G, Chen B L, Kuang X Y, Lu H Y, Lu C 2021 J. Phys. Chem. A 125 4126 doi: 10.1021/acs.jpca.1c02148 [31] Lu C, Gong W, Li Q, Chen C 2020 J. Phys. Chem. Lett. 11 9165 doi: 10.1021/acs.jpclett.0c02656 [32] Li W L, Chen T T, Chen W J, Li J, Wang L S, 2021 Nat. Commun. 12 6467 doi: 10.1038/s41467-021-26785-9 [33] Zhang J, Dolg M 2015 Phys. Chem. Chem. Phys. 17 24173 doi: 10.1039/C5CP04060D [34] Frisch M J, Trucks G W, Schlegel H B, et al. 2016 Gaussian 09 (Revision Ed. 01) (Wallingford, CT: Gaussian, Inc. [35] Lu T, Chen F W 2012 J. Comput. Chem. 33 580 doi: 10.1002/jcc.22885 [36] Humphrey W, Dalke A, Schulten K 1996 J. Mol. Graph. 14 33 doi: 10.1016/0263-7855(96)00018-5 [37] Dolg M, Stoll H, Savin A, Preuss H 1989 Theor. Chim. Acta. 75 173 doi: 10.1007/BF00528565 [38] Peterson A, Kirk F, Detlev G, Erich S H, Michael D 2003 J. Chem. Phys. 119 11113 doi: 10.1063/1.1622924 [39] Binkley, Stephen J, Pople, John A, Hehre, Warren J 1980 J. Am. Chem. Soc. 102 939 doi: 10.1021/ja00523a008 [40] Adamo C, Barone V 1999 J. Chem. Phys. 110 6158 doi: 10.1063/1.478522 [41] Wadt W R, Hay P J 1985 J. Chem. Phys. 82 284 doi: 10.1063/1.448800 [42] Krishnan R, Binkley J S, Seeger R, Pople J A 1980 J. Chem. Phys. 72 650 doi: 10.1063/1.438955 [43] Tozer D J, Handy N C 1998 J. Chem. Phys. 109 10180 doi: 10.1063/1.477711 [44] Alexandrova A N, Boldyrev A I, Zhai H J, Wang L S, Steiner E, Fowler P W 2003 J. Phys. Chem. A 107 1362 -
计量
- 文章访问数: 562
- HTML全文浏览数: 562
- PDF下载数: 7
- 施引文献: 0