
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
蛋白质作为基因功能的执行者,其表达模式和功能分析已成为后基因组学时代研究的核心内容之一。蛋白质组学是研究细胞、组织或器官内蛋白质整体表达的首选方法,也是研究蛋白质功能与相互作用的重要工具,其可深入揭示疾病机制或生理过程。自20世纪90年代以来,生物质谱技术迅速发展,由于准确性、灵敏度和高度自动化的优势,其已逐渐成为蛋白质组学研究的主要技术手段[1]。随着蛋白质组学研究的不断深入,研究重点从定性分析转为定量分析[2]。典型的蛋白质组学工作流程包括样品前处理、液相色谱(LC)分离、质谱(MS)检测分析、数据库搜索鉴定和对所获取的质谱数据进行下游分析[3]。
在2009年之前,常用于质谱检测的蛋白质样品制备方法包括凝胶内酶解[4-5]和溶液内酶解[6-7]。凝胶内酶解法是通过聚丙烯酰胺凝胶电泳(SDS-PAGE)分离不同分子质量的蛋白质,对凝胶脱色后,将其切成小块,进行还原烷基化二硫键、胰蛋白酶酶解蛋白成肽段,最后对肽段样品脱盐处理后进行质谱检测。该方法使用高效的阴离子表面活性剂十二烷基硫酸钠(SDS),可以溶解大多数蛋白质[8],但操作过程复杂、成本高、蛋白质损失大,且胶内酶解效率低、漏切率高。溶液内酶解法是直接在溶液中对蛋白质进行酶解,酶解前使用丙酮沉淀去除SDS等不利于质谱检测的表面活性剂。该方法酶解效率高、漏切率低,但耗时较长,丙酮沉淀蛋白后用高浓度尿素复溶易造成蛋白损失,且易引入聚合物造成质谱检测时产生信号干扰。
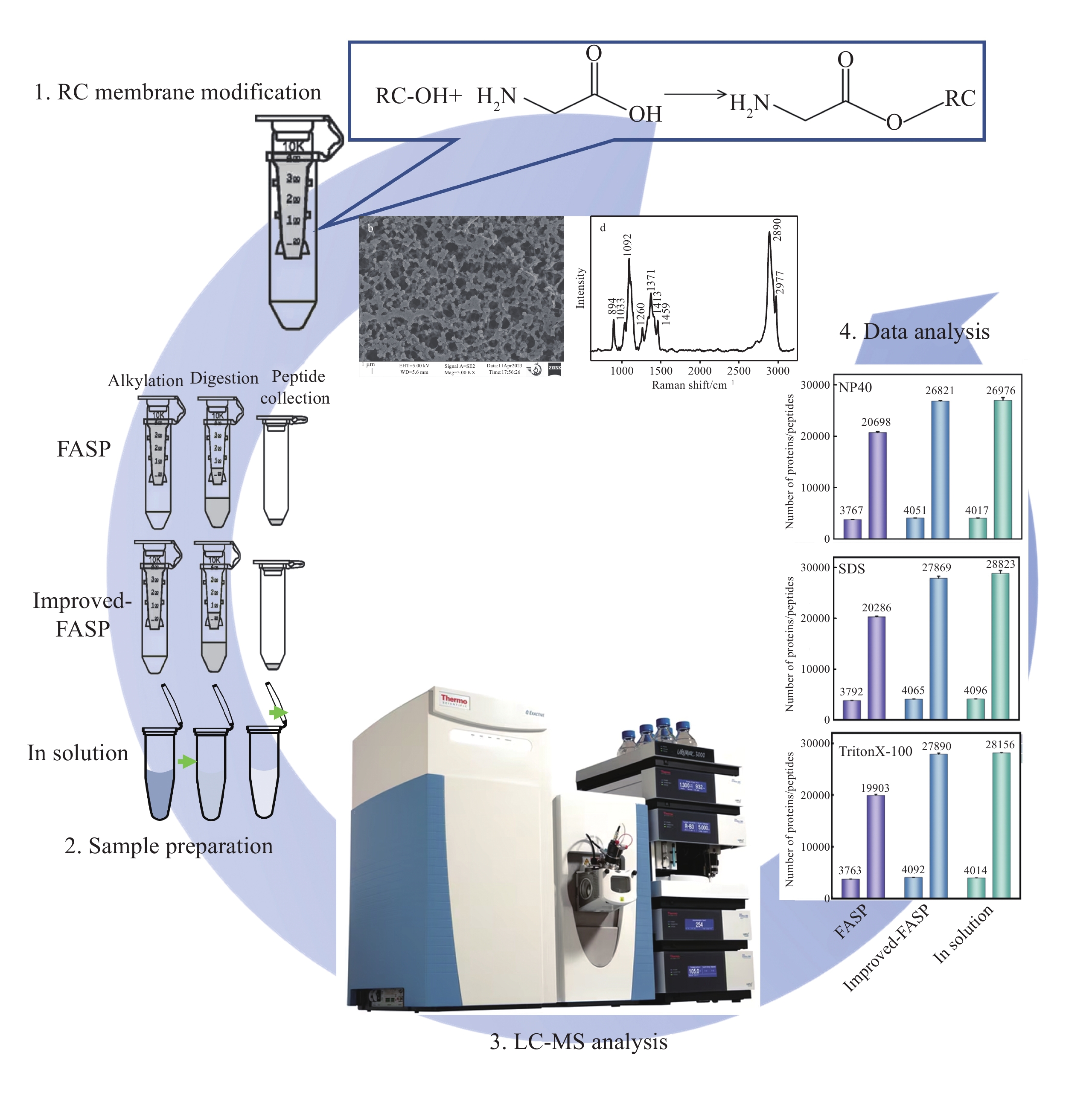
2009年,Wisniewski等[9]首次提出了一种新的蛋白质组学前处理方法——超滤辅助样品制备(filter-aided sample preparation,FASP),该方法是在超滤离心管的再生纤维素膜上进行一系列的还原烷基化和蛋白质酶解,通过超滤膜截留蛋白大分子,去除裂解液中的无机盐和表面活性剂等小分子,最后用NH4HCO3置换8 mol/L尿素缓冲液后进行蛋白质酶解,离心收集酶解肽段。该方法耗时短,样品污染小,避免了酶切不充分的蛋白质和过量胰酶对质谱鉴定的影响,同时克服了溶液内酶解法和胶内酶解法的缺陷,但存在超滤管本身对蛋白质的吸附导致肽段回收率降低的局限性。蛋白质吸附的原因是超滤膜表面的疏水性,这会导致蛋白质在膜表面的吸附量增加,进而使超滤膜截留的蛋白质增多,造成膜过滤阻力增大、通量衰减、分离效率降低。研究表明[10],增加膜表面亲水性会降低蛋白质在膜表面的吸附。
基于此,本研究提出一种改进后的FASP样品制备方法,通过对超滤膜表面进行接枝修饰,增强膜表面的亲水性,从而减少蛋白质吸附和膜污染,并以人宫颈癌细胞(HeLa)蛋白为研究对象,评估该方法的可行性。
-
Q Exactive™ Plus四极杆/静电场轨道阱高分辨质谱仪(配有纳升电喷雾离子源(Nano ESI)及Xcalibur 4.1数据采集系统)、Ultimate3000高效液相色谱仪、Proteome Discoverer 2.3数据处理软件、SpeedVac SPD1030离心浓缩仪、ESCALAB Xi+型X射线光电子能谱仪:美国Thermo Fisher Scientific公司产品;Milli-Q Integral10超纯水机:美国Millipore公司产品;KQ3200E超声波清洗仪:昆山舒美公司产品;TGL-16M低温高速离心机:湖南湘仪实验室仪器开发有限公司产品;InViaQontor激光拉曼光谱仪:英国雷尼绍公司产品;VERTEX70显微红外光谱仪:德国Bruker公司产品;Gemini SEM 500场发射扫描电子显微镜:德国蔡司公司产品;DSA100S接触角测量仪:德国KRUSS公司产品。
-
甲酸:质谱纯,美国赛默飞公司产品;甲醇、乙腈:色谱纯,德国密理博公司产品;测序级胰酶:生物试剂,美国Promega公司产品;NH4HCO3、尿素、二硫苏糖醇(DTT)、碘乙酰胺(IAA):纯度≥99%,德国Sigma公司产品;丙酮:上海Adamas试剂有限公司产品;三(羟甲基)氨基甲烷盐酸盐(Tris-HCl)、蛋白免疫印记和免疫沉淀(Western及IP)裂解液、SDS裂解液、RIPA裂解液(弱):生物试剂,碧云天生物技术有限公司产品;氢氧化钠:优级纯,天津福晨化学试剂有限公司产品;甘氨酸:合肥千盛生物科技有限公司产品;UFC5010 10 ku超滤离心管:美国密理博公司产品;HeLa 细胞:由西安交通大学生命学院提供。
-
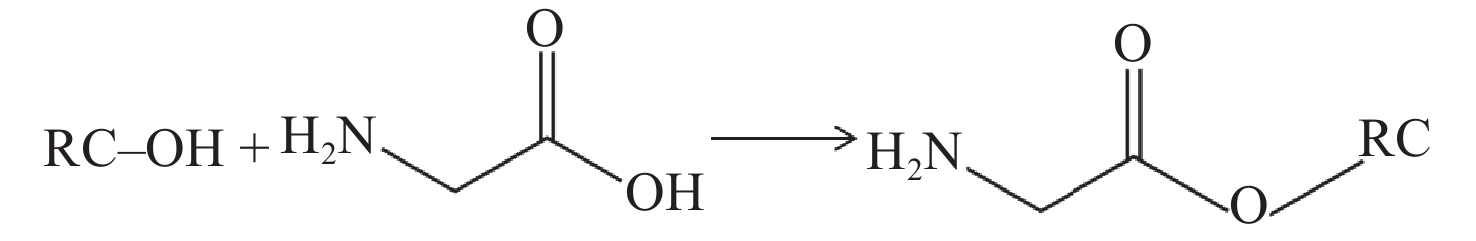
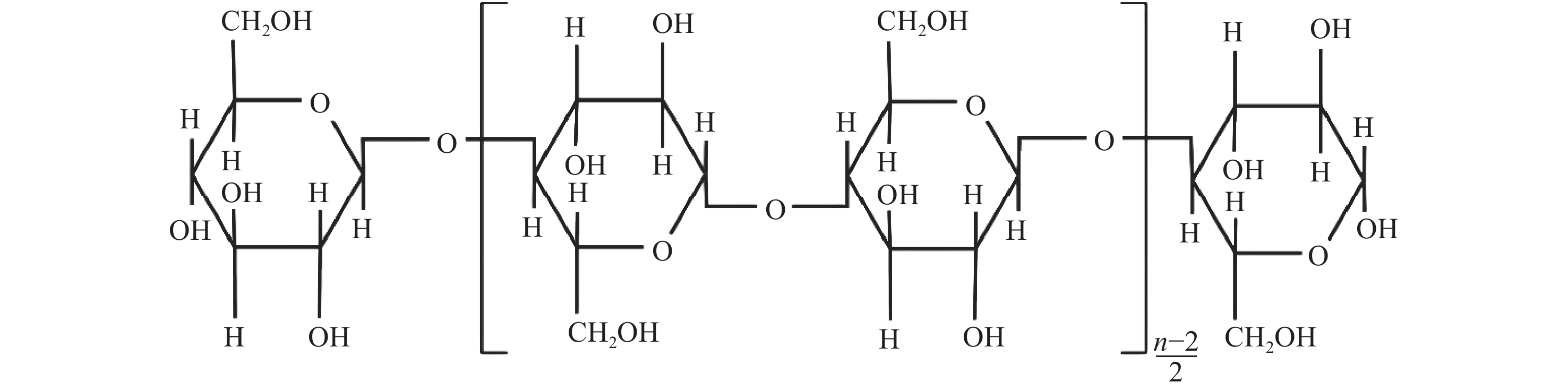
再生纤维素(regenerated cellulose,RC)超滤膜的核心成分是由D-葡萄糖酐通过1,4 β-型糖苷键连接而成的天然线型多糖高分子,其化学结构可表示为(C6H10O5)n,其中n代表聚合度,示于图1。
再生纤维超滤膜可以通过降低膜表面疏水性(即增加亲水性)来减少蛋白吸附,膜改性的有效方法之一是在纤维素分子上接入两性小分子,使亲水端朝外相互形成氢键,以提高亲水性。本研究选择空间位阻小的甘氨酸接入再生纤维素膜,示于图2。前期工作表明,直接接入甘氨酸时不易连接到膜表面且连接效率低。本研究对膜进行碱性活化后再接入甘氨酸,以提高连接效率。同时,为提升实验效果,需对超滤离心管中的商业RC膜进行预处理,以去除表面可能存在的甘油、防腐剂及润湿剂等添加剂。
原膜预处理:将RC膜置于异丙醇(IPA)中浸泡45~60 min,用超纯水冲洗,确保表面清洁无杂质。
原膜活化:将清洗干净的RC膜浸泡在6% NaOH溶液中1 h。
原膜改性:取出活化后的膜并冲洗干净,随后将其浸泡在1 mol/L甘氨酸(0.1 mol/L Tris-HCl)溶液中,45 ℃振荡反应12 h,干燥,备用。
-
采用激光拉曼光谱仪测试甘氨酸改性前后RC超滤膜表面的特征官能团,验证超滤膜表面接枝情况。选择激发波长532 nm、功率100 MW的激光器,设置拉曼光谱采集位移范围为3 200~100 cm−1。
-
采用显微红外光谱仪的透射(TR)模式测试甘氨酸改性前后RC超滤膜表面的特征官能团。设置红外光谱测试范围为4 000~400 cm−1。
-
利用XPS测试甘氨酸改性前后RC超滤膜表面元素组成及C、N、O元素的精细图谱,探究膜表面化学成分的改变。该仪器配备高强度的单色Al KαX 射线源,最佳能量分辨率可达≤0.45 eV。
-
甘氨酸改性前后RC超滤膜经离子溅射仪真空喷金处理后使其具有导电性,利用场发射扫描电子显微镜表征膜表面微观形貌,以分析改性处理对膜孔道结构的影响。
-
超滤膜的亲水性能可通过测量膜表面的水接触角大小来直观评估。本研究使用的接触角测量仪测量范围为0~180°,分辨率为0.01°,可通过瞬时捕捉图像记录水滴与膜表面的接触角。
-
采用牛血清蛋白(bovine serum albumin,BSA)的吸附效果来衡量超滤膜对蛋白质的吸附量。配制1 g/L BSA溶液,将RC原膜和改性后的RC膜(1 cm×1.5 cm)完全浸泡在1 g/L BSA溶液中24 h,以11 200 r/min离心15 min,收集流出液,使用二辛可酸(BCA)法测量BSA蛋白含量,按照式(1)计算膜表面的吸附量。
式中,q为蛋白吸附量;C0为吸附前BSA浓度;C1为吸附后BSA浓度;V为BSA溶液体积;m为膜表面积。
-
在6孔板中培养人宫颈癌HeLa细胞至密度80%~90%时收集细胞,加入200 μL裂解液,吹打混匀后裂解10 min,以11 200 r/min离心5 min,取上清液,采用BCA法测量蛋白浓度。
取20 μg蛋白裂解液,加入200 mL UA缓冲液(8 mol/L尿素,0.1 mol/L Tris-HCl(pH 8.5))以及2.3 μL 1 mol/L DTT,37 ℃孵育1~2.5 h,转移至10 ku超滤离心管,以11 200 r/min离心20 min,加入200 μL UA重复离心并弃去下部收集管中的流出液。随后向上部超滤管中加入100 μL UA溶液(含50 mmol/L IAA),避光孵育40 min,离心弃去流出液,重复加入200 μL UA,离心洗涤1次。最后加入200 μL 50 mmol/L NH4HCO3溶液,离心洗涤2次,弃去流出液和旧的收集管,更换新的收集管,向上部超滤管中加入100~200 μL含胰酶(胰酶:蛋白=1:25~1:20(W/W))的50 mmol/L NH4HCO3溶液,37 ℃孵育过夜,离心后收集下部收集管中肽段液体,再向超滤管中加入200 μL 50 mmol/L NH4HCO3,离心洗涤1次,合并2次离心后收集管中的肽段溶液。除盐柱脱盐后,以20 μL 0.1%甲酸溶解肽段样品,进行质谱分析。
-
向1.5 mL带盖离心管中加入20 μg HeLa细胞蛋白,并用50 mmol/L NH4HCO3(含8 mol/L尿素)补足至50 μL,加入1 mol/L DTT还原,使DTT终浓度为10 mmol/L,37 ℃孵育1~2.5 h。随后,加入500 mmol/L IAA,使IAA终浓度为50 mmol/L,室温避光孵育40 min后,加入6倍体积的丙酮(−20 ℃预冷)沉淀蛋白,扣紧离心管盖后缓慢颠倒样品3次使溶液充分混匀,将沉淀的样品放入−20 ℃冰箱中静置4~15 h或过夜,使蛋白完全沉淀。沉淀完成后,恢复至4 ℃,使尿素完全溶于丙酮,以10 000 r/min离心10 min,弃去上清液。加入1 mL 90%丙酮(−20 ℃预冷),扣紧盖子后缓慢颠倒样品3次,以10 000 r/min离心10 min,弃去上清液(重复1次)。待管中蛋白沉淀丙酮完全挥干后,加入50 μL 100 mmol/L NH4HCO3(含6 mol/L尿素),涡旋3 min溶解蛋白,加入50 μL水,混合均匀。按照胰酶:蛋白=1:25~1:20(W/W)加入胰酶,37 ℃孵育过夜,随后用离心浓缩仪抽干液体,终止酶切。肽段用Ziptip C18除盐柱除盐,干燥后用20 μL 0.1%甲酸复溶,进行质谱分析。
-
C18分析柱(150 mm×150 µm,1.9 µm),C18 trap柱(20 mm×75 µm,3 µm);流动相:0.1%甲酸水溶液(A)和0.1%甲酸-80.0%乙腈溶液(B);梯度洗脱:0~7.0 min(3.0%B),7.0~10.0 min(3.0%~15.0%B),10.0~62.0 min(15.0%~29.0%B), 62.0~80.0 min(29.0%~41.0%B),80.0~85.0 min(41.0%~100.0%B),85.0~90.0 min(100.0%B),90.0~90.1 min(100.0%~3.0%B);流速0.6 µL/min;进样量2 µL。
-
正离子模式,喷雾电压2.0 kV,毛细管温度275 ℃,离子透镜射频电压(S-Lens RF)55%,一级分辨率35 000,二级分辨率17 500,自动增益控制目标(AGC target)一级设为 3×106、二级设为 1×106,最大驻留时间(Maximum IT)50 ms,碰撞能量27,固定第一质量谱(Fixed first mass)m/z 200,一级质量扫描范围m/z 350~1 200。采用数据依赖性扫描(DDA)模式: 设置一级母离子全扫描和数据依赖的二级子离子扫描(Full MS/dd-MS2)扫描类型,隔离窗口m/z 1.6,最高强度子离子数(TopN)为10。
-
将质谱3次采集的原始数据RAW文件导入Proteome Discoverer 2.3软件进行蛋白搜库鉴定(人蛋白数据库,human;Uniprot Human_9606;Homo_sapiens_9606),采用默认参数。设置母离子质量偏差10−5;碎片离子质量偏差0.020 u;固定修饰:半胱氨酸烷基化(+57.021 u);动态修饰:甲硫氨酸氧化(+15.995 u);天冬酰胺和谷氨酰胺脱氨基化(+0.984 u);trypsin漏切位点0;假阳性发现率(FDR)<0.01。搜库结束后,从结果中导出.pdResult和.fasta源文件保存。
-
为验证膜表面已成功接枝氨基酸基团,本研究采用激光共聚焦拉曼光谱仪检测膜表面的官能团结构,同时在电镜下观察膜表面的结构变化。改性前后膜表面在电镜下的结构特征分别示于图3a、3b,未改性和甘氨酸改性后2种RC膜表面的拉曼光谱特征分别示于图3c、3d。结果表明,改性后RC超滤膜表面未见明显的结构变化,RC膜孔径大小未发生明显变化,拉曼光谱新增1 033 cm−1处C−N含氮官能团和1 413 cm−1处−COO−含氧官能团。
-
利用XPS能谱技术进一步探究改性后RC膜表面的氮元素含量。未改性膜和甘氨酸改性膜的XPS全谱示于图4,均主要含有C、O骨架元素。同时,在甘氨酸改性膜的XPS全谱峰图中,除主要的C、O元素外,还存在微量的N元素,这是由甘氨酸分子NH2基团引入的,表明甘氨酸分子被连接到RC膜表面。
使用Avantage软件进一步对C、O、N元素进行分峰拟合高分辨分析,为确保数据的准确性,以外来污染碳的峰(284.8 eV)作为基准进行精细荷电校正,消除荷电效应对结合能测定的影响,保证分析结果的可靠性。通过C1s、O1s和N1s谱分析C、N、O元素在改性RC膜表面的化学状态,改性前后2种RC膜表面C1s和O1s分峰拟合高分辨图示于图5。从图5a的C1s分峰拟合结果发现,未改性RC膜表面存在2种主要的碳化学键合状态,即结合能284.8 eV的C−H键和结合能286.3 eV的C−O−C/C−OH键[9],表明在未改性膜中C主要与H、O形成化学键;而对于甘氨酸改性后的RC膜,观察到C1s分峰拟合出现结合能为286.4 eV和288.7 eV的2个新峰,分别归属为C−N和O−C=O键[10],来源于甘氨酸分子结构。此外,在图5b的O1s分峰拟合图中,甘氨酸改性RC膜新增了1个结合能533.0 eV的峰,归属为C=O键[11]。上述结果均印证了改性后膜表面引入了甘氨酸分子的化学键合状态,证明甘氨酸成功连接至RC膜表面。
-
本实验比较了改性前后2种RC膜表面接触角的变化,示于图6。其中,未改性膜表面平均接触角为36.8°,经甘氨酸改性后,接触角平均值下降至4.8°。结合1.3节,证实了引入甘氨酸后膜表面化学组成和结构的改变使膜表面的亲水性增强,导致接触角减小。
改性前后2种RC膜表面水接触角随时间的变化示于图7。改性后的RC膜初始水接触角为25.3°,但随着时间推移,接触角降至4.8°;而未改性处理的RC膜水接触角由初始的55.57°最终降至36.8°,降低幅度较小。表明改性处理可以提高RC膜表面的亲水性。
-
对未改性膜和甘氨酸改性RC膜表面蛋白吸附量进行测试,其BSA标准蛋白吸附量分别为4.6、2.8 μg/cm2。可知,经改性处理的膜表面蛋白吸附量降低,表明膜表面抗蛋白黏附性能提高。
-
用改性后的RC超滤膜进行蛋白FASP样品前处理(改进FASP),并与经典FASP方法、溶液内酶解制样方法比较。同时,考察3种不同表面活性剂成分裂解液提取蛋白对后续制样效果的影响。
在3种方法中,HeLa蛋白处理量20 μg,进行3次重复实验,质谱进样量2 μL。经3次质谱DDA数据采集后,利用Proteome Discoverer 2.3软件对原始数据进行搜库鉴定和定量分析,最终蛋白和肽段的鉴定结果示于图8。
NP40裂解液提取蛋白后,改进FASP方法比FASP方法鉴定到的蛋白数量增加约7.54%,肽段数量增加约29.58%,示于图8a。SDS裂解液提取蛋白后,改进FASP方法比FASP方法鉴定到的蛋白数量增加约7.20%,肽段数量增加约37.38%,示于图8b。TritonX-100提取蛋白后,改进FASP方法比FASP方法鉴定到的蛋白数量增加约8.74%,肽段数量增加约40.13%,示于图8c。
在不同样品前处理方法下,质谱定量检测到的肽段丰度分布示于图9。可以看出,3种不同裂解液提取蛋白后,与经典FASP方法相比,改进FASP方法检测到丰度大于104的肽段数量明显增多,丰度小于104的肽段数量明显减少。表明改进FASP方法能够获得更多的肽段,从而增强低丰度肽段的信号响应。此外,与溶液内酶解制样相比,改进FASP方法能检测到更多丰度大于106的肽段,且重现性更好。改进FASP方法鉴定到的蛋白和肽段数量比FASP方法明显增多,与溶液内酶解制样方法相当。
不同裂解液中3种样品前处理方法蛋白等电点分布以及蛋白分子质量分布分别示于图10、11。可见,改进FASP方法鉴定到各等电点和分子质量分布的蛋白数量均比FASP方法多,表明该方法可以稳定提升鉴定到的蛋白数量。
-
在FASP方法制备样品过程中,蛋白柱载量的优化对于提高蛋白质分析的灵敏度和准确性至关重要。因此,本实验在保持LC-MS进样量2 μL的条件下,考察不同蛋白柱载量。选择相同的Western及IP裂解液(裂解成分1% TritonX-100)提取蛋白,分别在超滤管中处理5、10、20、40和60 μg HeLa细胞蛋白,为确保结果的可靠性和稳定性,每个柱载量均进行3次重复实验。不同起始蛋白处理量的最终上样量在0.3~2.5 μg之间,以保证不超过色谱分析柱的最大柱载量。不同蛋白处理量蛋白和肽段鉴定结果示于图12。可见,改性后的RC膜处理蛋白的最佳柱载量为20 μg(0.5 mL,3 ku截留分子质量(MWCO)),该处理量足以进行后续的质谱分析。
-
本研究基于亲水改性超滤膜提出了一种改进FASP制样方法。使用改进FASP蛋白前处理方法结合超快速高分辨质谱,以HeLa细胞为研究对象进行LC-MS/MS检测,并比较FASP法、改进FASP法、溶液内酶解法鉴定蛋白质和肽段数量的差异。结果表明,3种不同裂解液对改进FASP方法均适用,采用改进FASP方法鉴定的蛋白质和肽段数量比FASP方法分别增加了约7%~9%、30%~40%,与溶液内酶解法相当。同时,改进FASP法检测到丰度大于104的肽段数量比FASP方法多,而且比溶液内酶解方法检测到更多丰度大于106的肽段,重现性更好。此外,改进FASP方法鉴定到的蛋白分子质量和等电点分布无偏向性,具有通用性。最后,确定了改进FASP方法的最佳蛋白柱载量为20 μg,并且微量蛋白样品(5 μg)也可使用该方法进行样品前处理。
结果表明,通过接枝两性小分子提高再生纤维素膜亲水性后,可以降低膜对蛋白样品的吸附作用,提高蛋白和肽段的鉴定数量。该方法提升了蛋白样品的制样效果,尤其对于珍贵的微量样品,在样品前处理步骤减少了蛋白损失,有助于鉴定更多的蛋白和肽段。此方法有望为疾病相关生物标志物筛选,药物药效机制研究,效应靶点鉴定以及疾病诊断、监测和治疗等领域提供帮助。
基于高分辨质谱的超滤辅助样品制备方法的优化
Optimization of Filter-aided Sample Preparation Method Based on High-resolution Mass Spectrometry
-
摘要: 超滤辅助样品制备(filter-aided sample preparation,FASP)法广泛用于蛋白质样品前处理,但该方法使用的超滤膜会非特异性吸附蛋白质而造成样品损失。本研究提出了一种优化后的FASP制样方法,通过对超滤膜表面接枝两性氨基酸的化学修饰方法,提高超滤膜表面亲水性,以降低蛋白质损失。利用优化的FASP方法制备人宫颈癌细胞(HeLa)蛋白样品,并进行LC-MS/MS检测,比较了FASP方法、改进后FASP方法、溶液内酶解法鉴定到的肽段和蛋白质数量。结果表明,改进后的FASP方法鉴定到的蛋白质和肽段数目均有所提高,分别增加了7%~9%、30%~40%。同时,确定了改进后FASP方法的最佳蛋白柱载量为20 µg。
-
关键词:
- 超滤辅助样品制备(FASP) /
- 蛋白质样品前处理 /
- 亲水改性 /
- 高分辨质谱 /
- 蛋白质组学
Abstract: This study centered on an improved filter-aided sample preparation (FASP) method for proteomic sample preparation. The main strategy focused on changing the chemical and physical properties of regenerated cellulose (RC) membranes in ultrafiltration tubes to reduce the loss of protein sample due to non-specific absorption on the membranes. Firstly, the RC membrane was immersed in isopropanol for the activation, followed by immersion in a 6% NaOH solution for the alkalization. Then, the amphiphilic small molecule glycine was grafted onto the surface of the activated RC membrane to enhance its hydrophilicity. The regenerated ultrafiltration membrane was characterized from multiple perspectives. The electron microscopy showed that the membrane structure and pore size remained unchanged after modification, the Raman spectroscopy coupled with X-ray photoelectron spectroscopy (XPS) demonstrated that glycine was successfully grafted onto the surface of the regenerated cellulose membrane, the hydrophilicity test indicated a decrease in the contact angle of the modified membrane, suggesting enhanced hydrophilicity. The protein adsorption test showed a reduction in bovine serum albumin (BSA) protein adsorption on the modified membrane, thus minimizing protein sample loss. When using this improved FASP method with Q Exactive™ Plus for LC-MS/MS analysis of HeLa cells, compared with the traditional FASP method and the in-solution digestion method, it was found that the improved FASP method, which is applicable to three types of lysis buffers (NP40, SDS, and Triton X-100), can increase the number of identified proteins by approximately 7%-9% and the number of identified peptides by approximately 30%-40%. Besides, the improved FASP method also has advantages in terms of peptide abundance distribution. For instance, it can detect more peptides with an abundance greater than 104 compared with the traditional FASP method and more peptides with an abundance greater than 106 compared with the in-solution digestion method, along with better reproducibility. Moreover, the improved FASP method shows no bias for identifying proteins with different molecular weights and isoelectric points, and its optimal protein loading is determined to be 20 μg. The developed method can reduce protein loss during sample preparation, especially for the precious trace samples. This is helpful for screening disease biomarkers, exploring drug efficacy mechanisms, identifying effective targets, and promoting the diagnosis, monitoring, and treatment of diseases. However, as the study did not clearly specify the limitations of this method, further exploration can be conducted to assess whether this method can be applied to different types of cells or more complex disease research. -

-
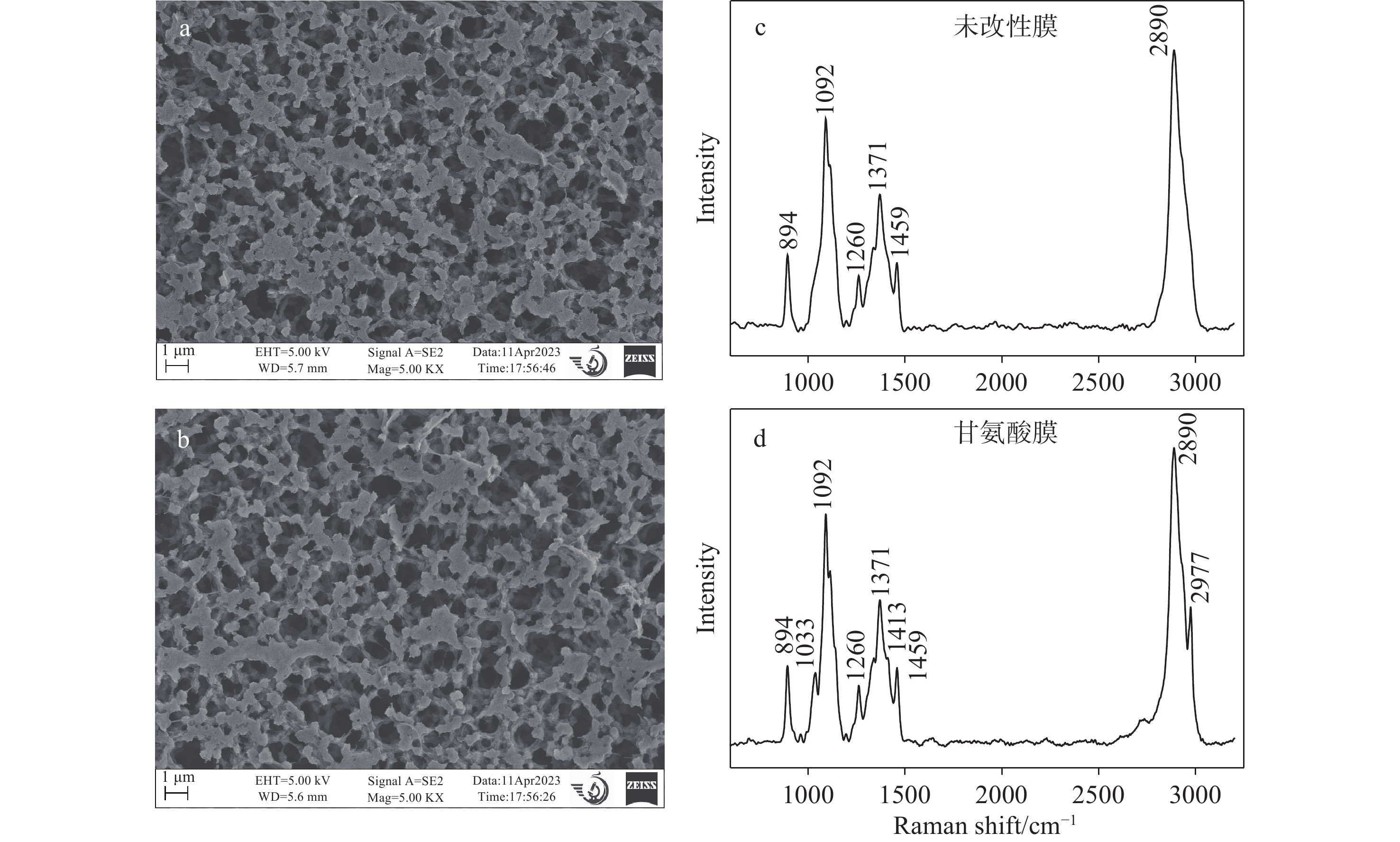
图 3 膜改性前后电镜(a, b)和拉曼光谱(c,d)特征
Figure 3. Electron microscopy (a, b) and Raman spectroscopy (c, d) characteristics of the modified and unmodified RC membranes
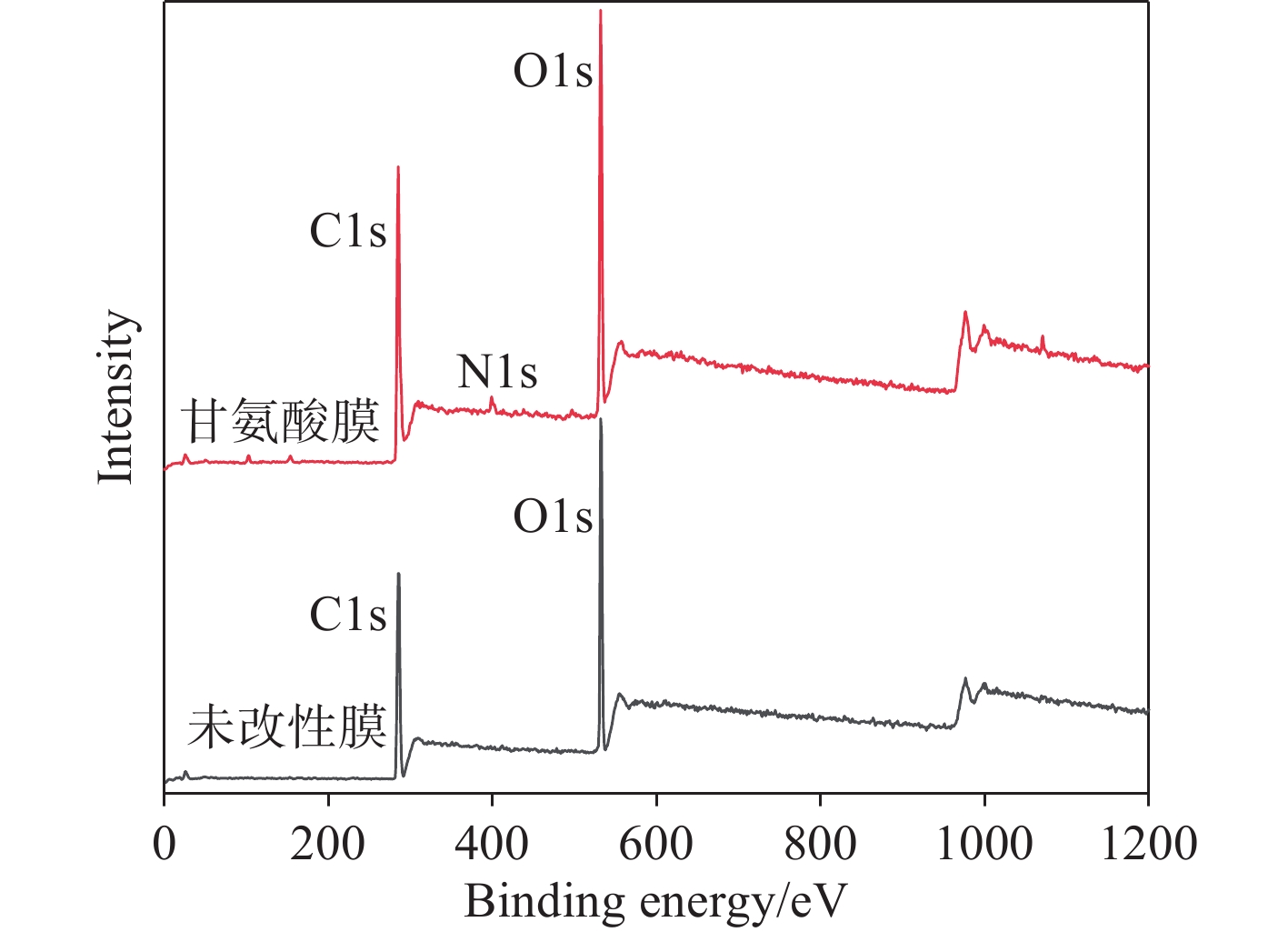
图 4 改性前后RC膜的XPS全谱图
Figure 4. XPS survey spectra of the modified and unmodified RC membranes
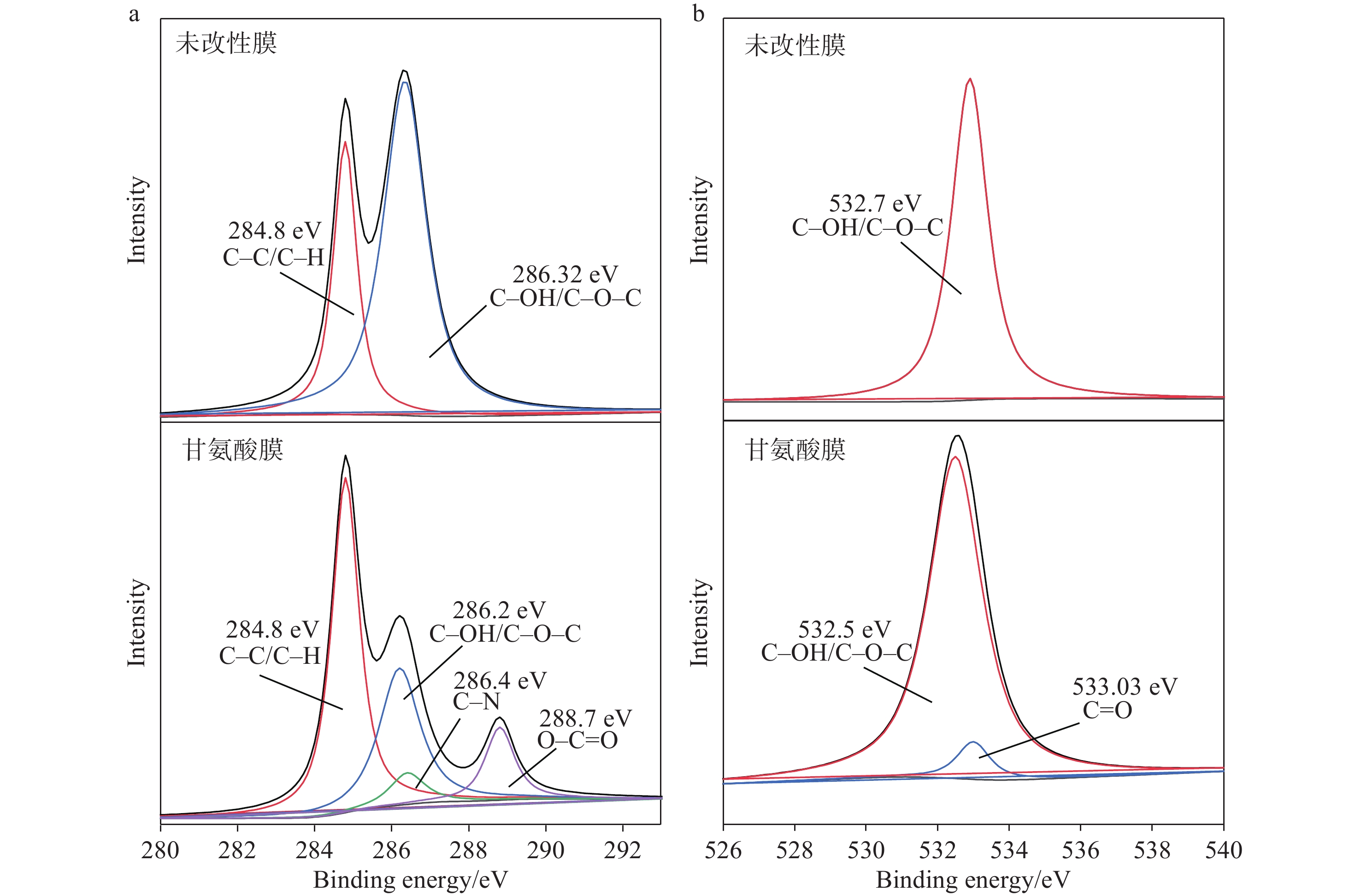
图 5 改性前后RC膜的C1s (a)和O1s (b)XPS高分辨图
Figure 5. High-resolution XPS spectra of C1s (a) and O1s (b) for the modified and unmodified RC membranes
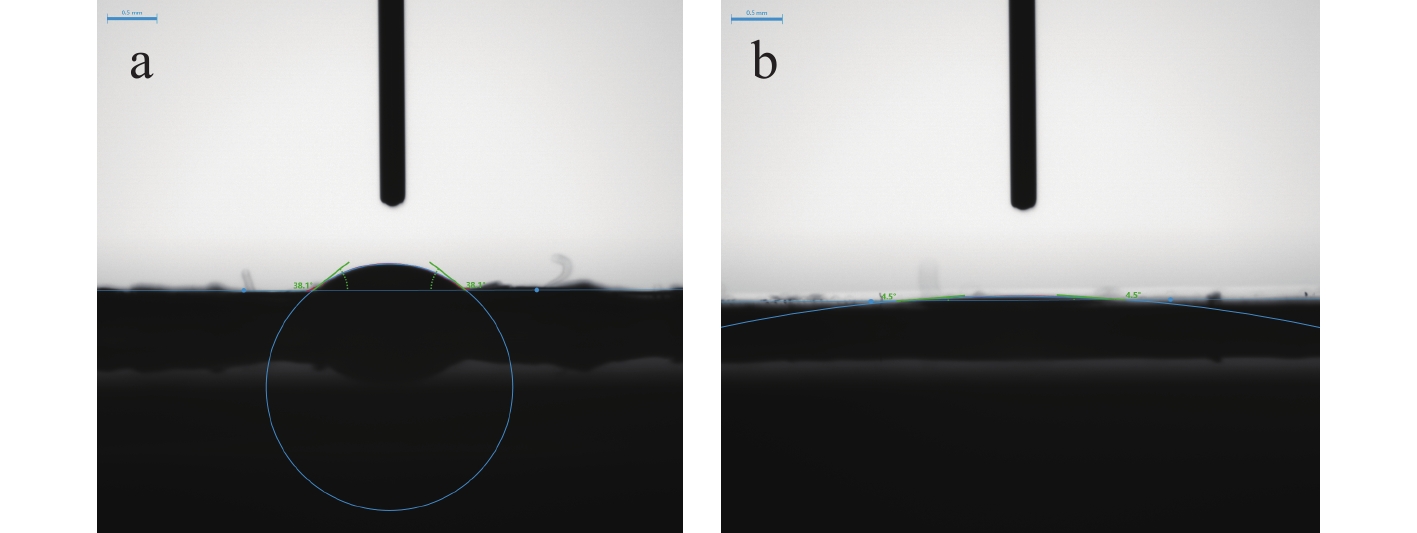
图 6 改性前(a)后(b)膜表面接触角测量
Figure 6. Surface contact angle measurements of the membrane before (a) and after (b) glycine modified
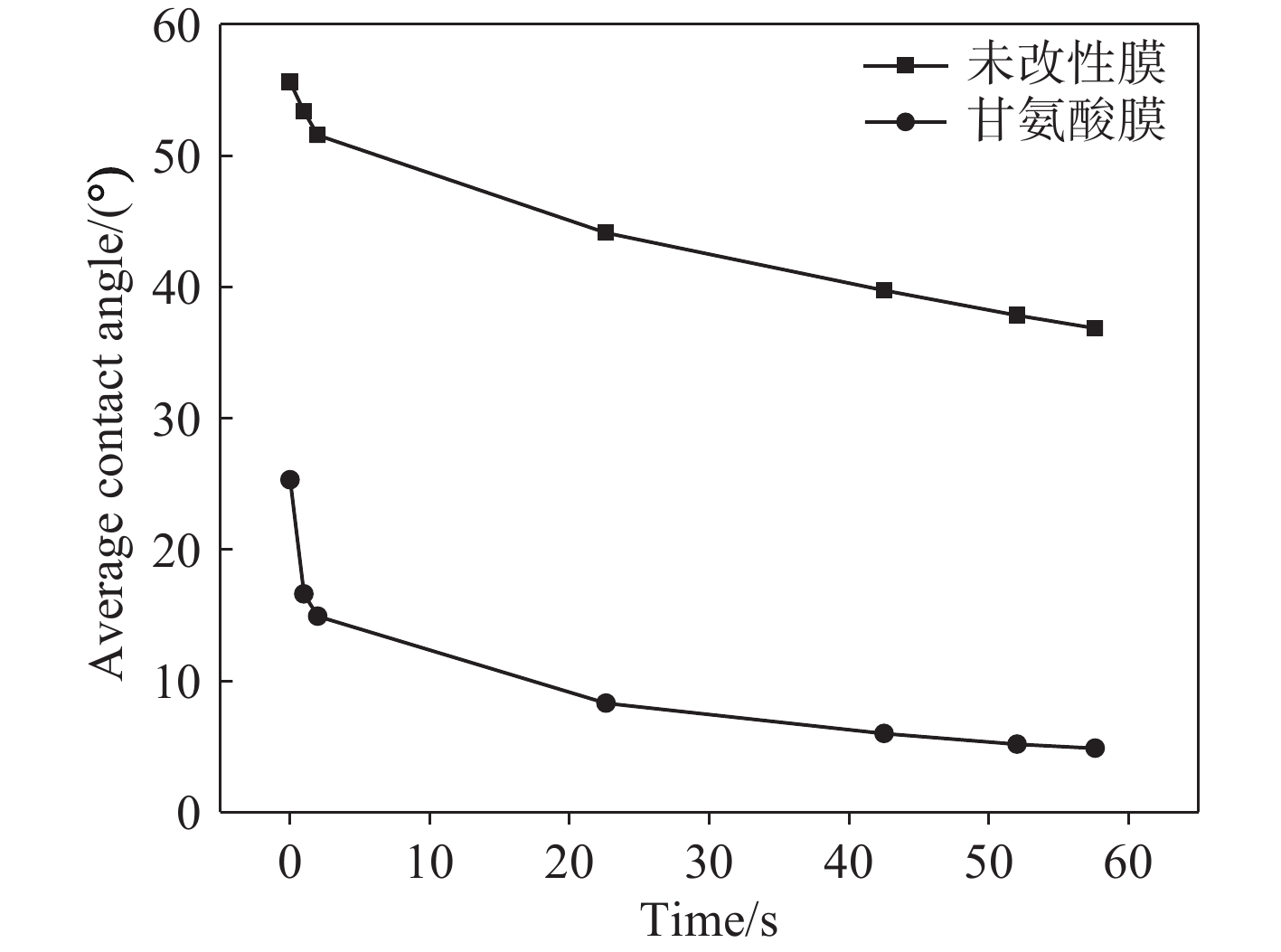
图 7 改性前后RC膜接触角变化
Figure 7. Changes in contact angle of the unmodified and modified RC membranes
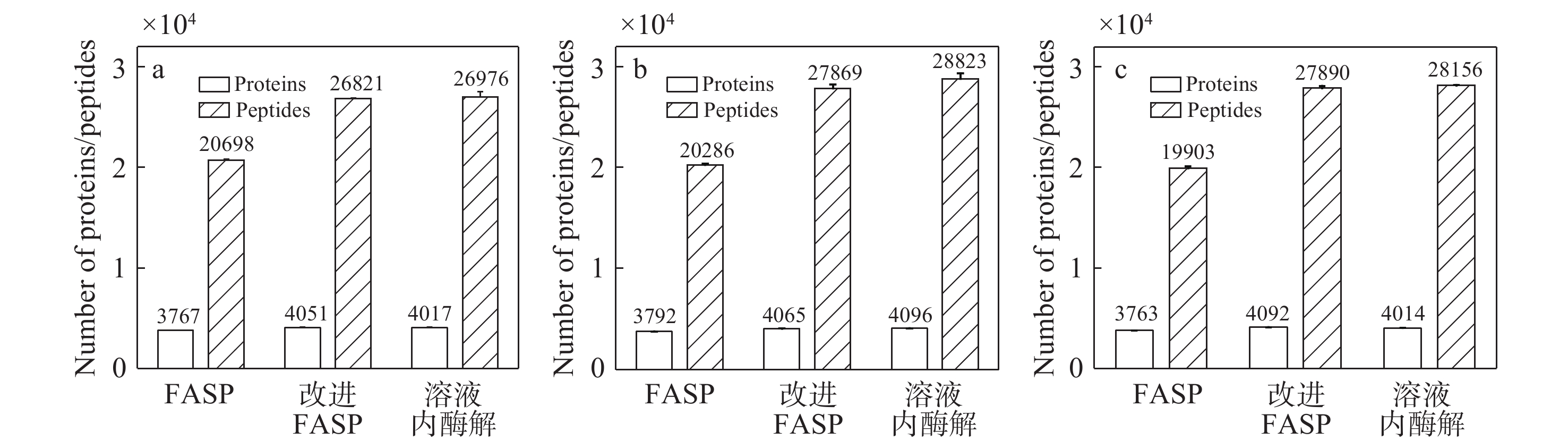
图 8 不同裂解液中3种样品前处理方法的质谱鉴定结果
Figure 8. Identification results of proteins and peptides by LC-MS using three sample preparation methods in different lysis buffers
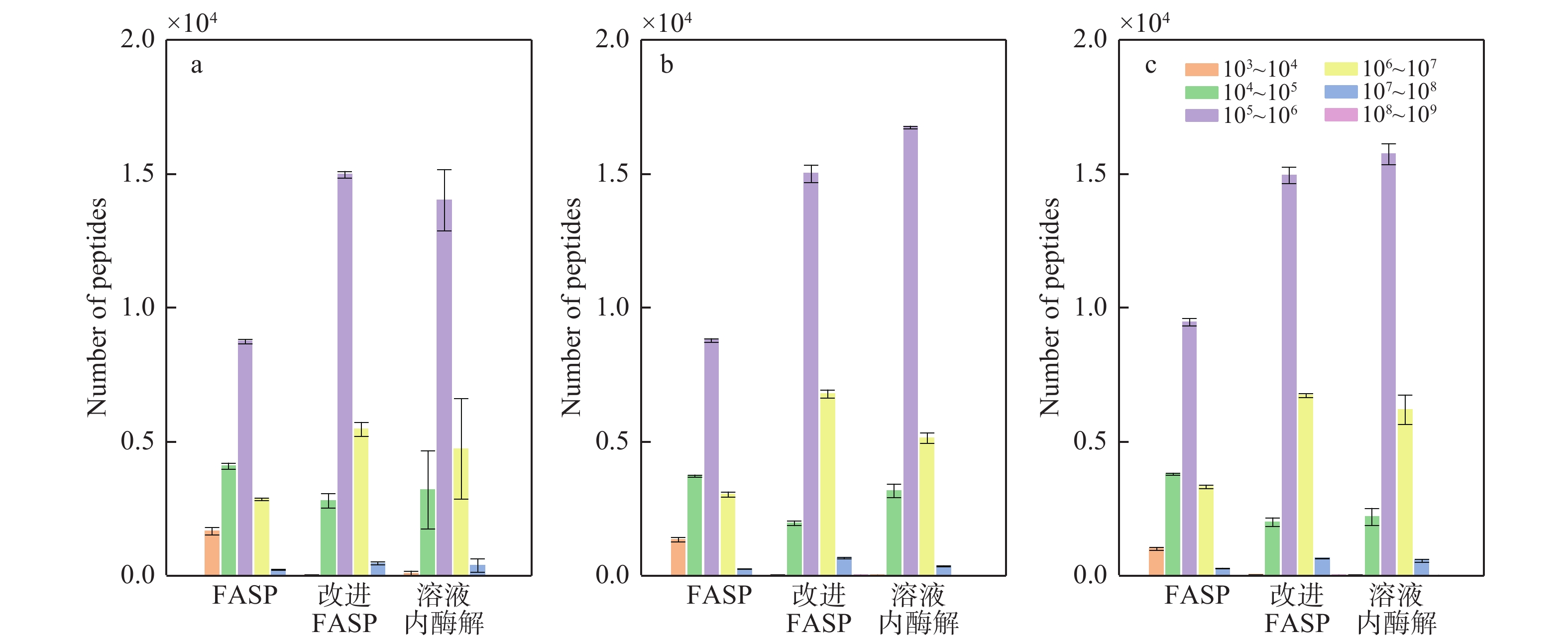
图 9 不同裂解液中3种样品前处理方法质谱检测肽段丰度分布
Figure 9. Distributions of peptide abundance by LC-MS using three sample preparation methods in different lysis buffers
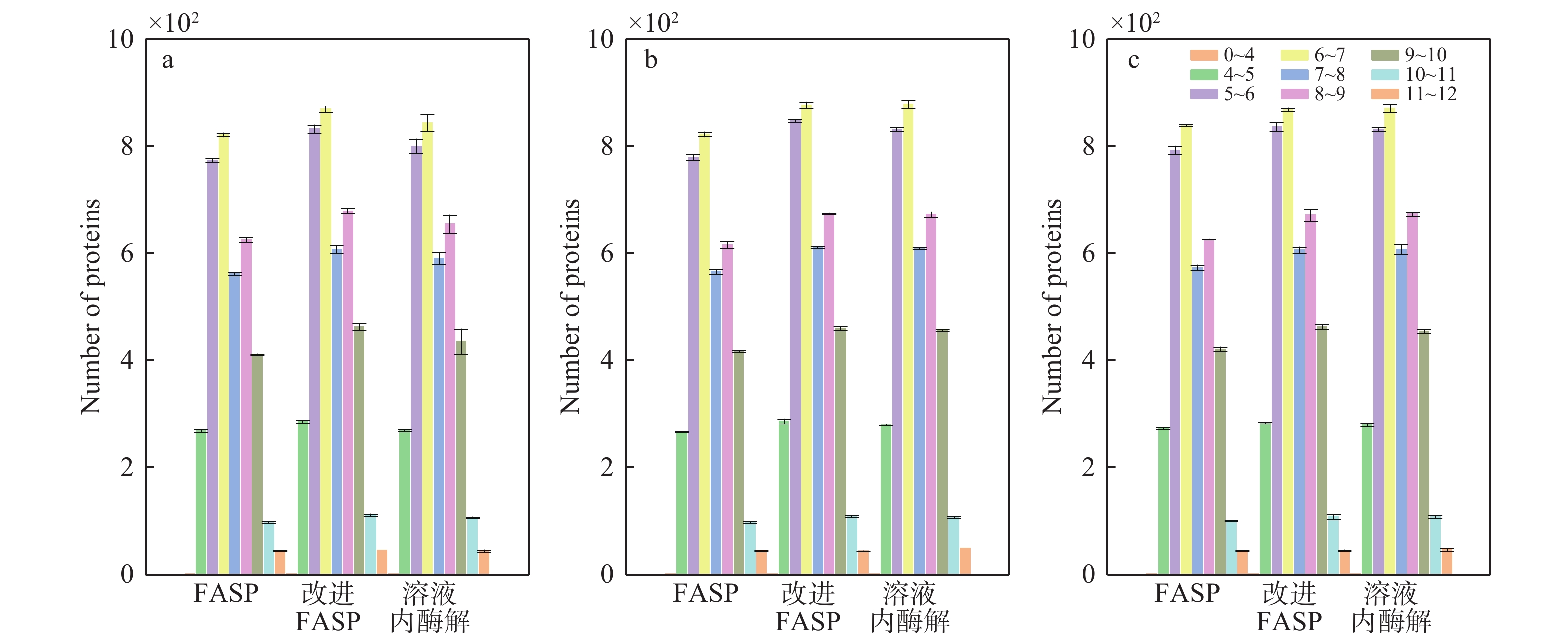
图 10 不同裂解液中3种样品前处理方法鉴定蛋白等电点分布
Figure 10. Isoelectric point distributions of proteins using three sample preparation methods in different lysis buffers
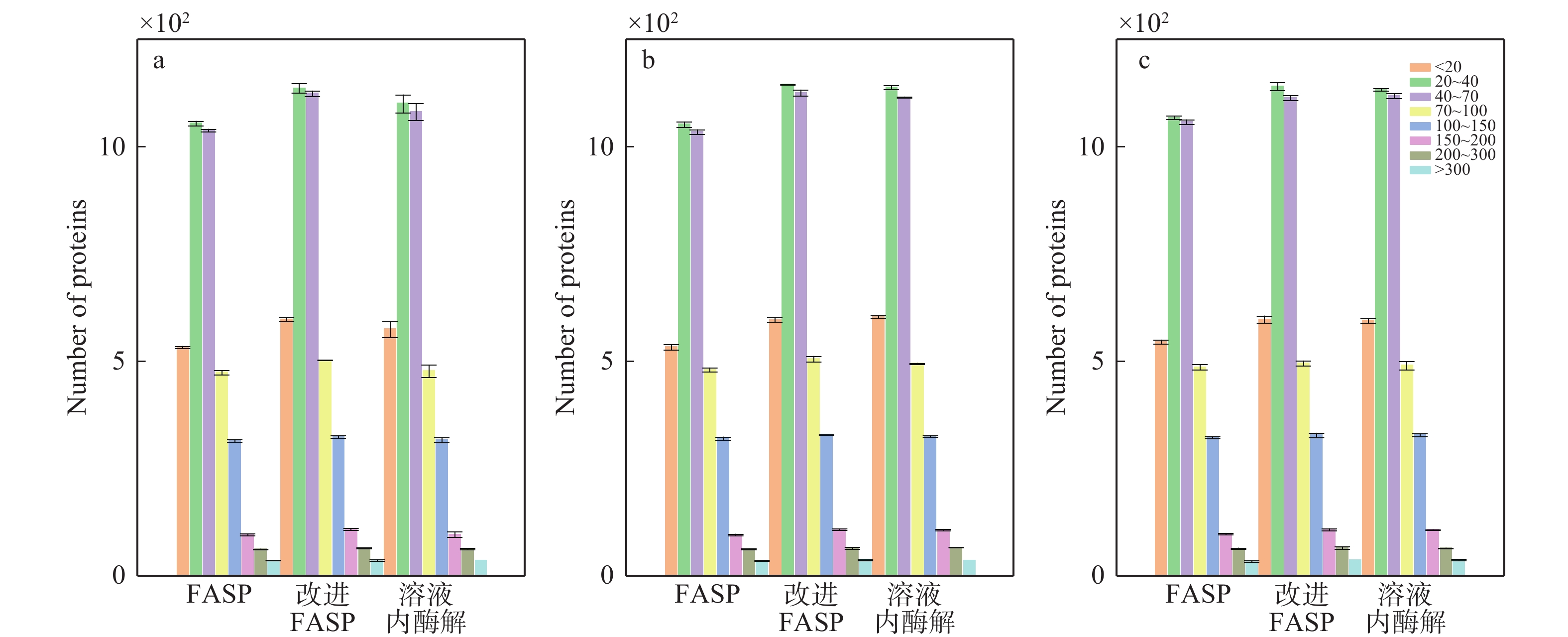
图 11 3种样品前处理方法鉴定蛋白分子质量分布
Figure 11. Molecular weight distributions of identified proteins in three sample preparation methods
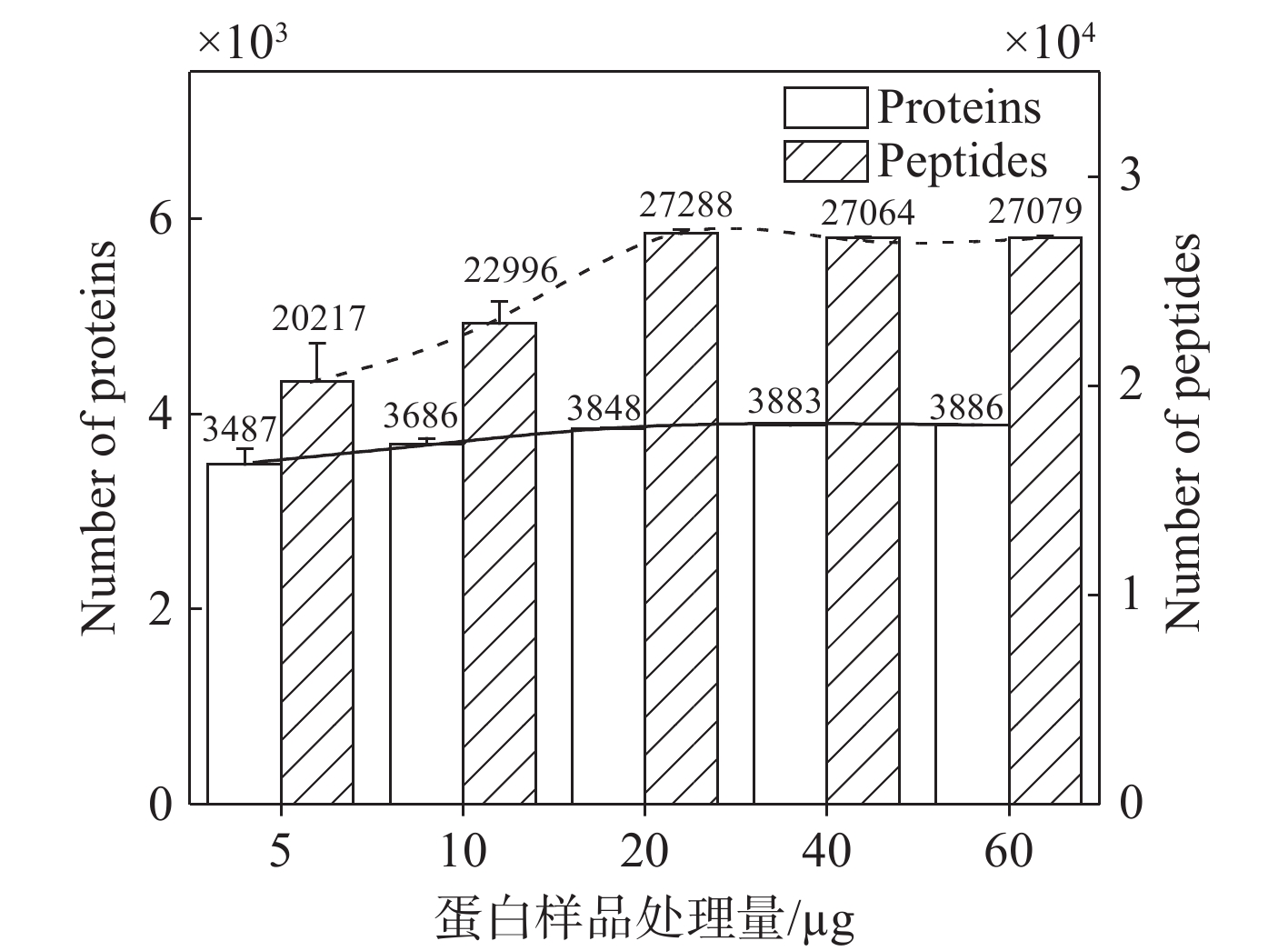
图 12 不同蛋白处理量蛋白和肽段鉴定结果
Figure 12. Number of proteins and peptides identified in different amounts of protein preparation
-
[1] WILKINS M R, PASQUALI C, APPEL R D, OU K, GOLAZ O, SANCHEZ J C, YAN J X, GOOLEY A A, HUGHES G, HUMPHERY-SMITH I, WILLIAMS K L, HOCHSTRASSER D F. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and arnino acid analysis[J]. Nature Biotechnology, 1996, 14(1): 61 -65 . doi: 10.1038/nbt0196-61[2] van der SPEK S J F, KOOPMANS F, PALIUKHOVICH I, RAMSDEN S L, HARVEY K, HARVEY R J, SMIT A B, LI K W. Glycine receptor complex analysis using immunoprecipitation-blue native gel electrophoresis-mass spectrometry[J]. Proteomics, 2020, 20(3/4): 1 900 403. [3] LI K W, CHEN N, KLEMMER P, KOOPMANS F, KARUPOTHULA R, SMIT A B. Identifying true protein complex constituents in interaction proteomics: the example of the DMXL2 protein complex[J]. Proteomics, 2012, 12(15/16): 2 428-2 432. [4] WU C C, MacCOSS M J, HOWELL K E, YATES J R. A method for the comprehensive proteomic analysis of membrane proteins[J]. Nature Biotechnology, 2003, 21(5): 532 -538 . doi: 10.1038/nbt819[5] BLONDER J, XIAO Z, VEENSTRA T D. Proteomic profiling of differentiating osteoblasts[J]. Expert Review of Proteomics, 2006, 3(5): 483 -496 . doi: 10.1586/14789450.3.5.483[6] NAGARAJ N, LU A, MANN M, WIŚNIEWSKI J R. Detergent-based but gel-free method allows identification of several hundred membrane proteins in single LC-MS runs[J]. Journal of Proteome Research, 2008, 7(11): 5028 -5032 . doi: 10.1021/pr800412j[7] WIŚNIEWSKI J R, ZOUGMAN A, MANN M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome[J]. Journal of Proteome Research, 2009, 8(12): 5 674-5 678. [8] 卞晓锴, 陆晓峰, 施柳青. 蛋白质超滤过程及超滤膜的表面改性研究现状[J]. 膜科学与技术, 2001, 21(4): 46 -51 . doi: 10.3969/j.issn.1007-8924.2001.04.011 BIAN Xiaokai, LU Xiaofeng, SHI Liuqing. The progress status of protein ultrafiltration and surface modification of ultrafiltration membranes[J]. Membrane Science and Technology, 2001, 21(4):46 -51 (in Chinese). doi: 10.3969/j.issn.1007-8924.2001.04.011[9] YOU J, ZHAO C, CAO J, ZHOU J, ZHANG L. Fabrication of high-density silver nanoparticles on the surface ofalginate microspheres for application in catalytic reaction[J]. Journal of Materials Chemistry A, 2014, 2(22): 8 491-8 499. [10] SHCHUKAREV A V, KOROLKOV D V. XPS study of group IA carbonates[J]. Central European Journal of Chemistry, 2004, 2(2): 347 -362 .[11] NOHIRA H, TSAI W, BESLING W, YOUNG E, PETRY J, CONARD T, VANDERVORST W, de GENDT S, HEYNS M, MAES J, TUOMINEN M. Characterization of ALCVD-Al2O3 and ZrO2 layer using X-ray photoelectron spectroscopy[J]. Journal of Non-Crystalline Solids, 2002, 303(1): 83 -87 . doi: 10.1016/S0022-3093(02)00970-5 -
计量
- 文章访问数: 59
- HTML全文浏览数: 59
- PDF下载数: 0
- 施引文献: 0