
 首页
首页 登录
登录 注册
注册





HTML
-
Electron momentum spectroscopy (EMS), alternatively referred to as binary (e, 2e) spectroscopy, has a distinctive ability in measuring orbital-resolvable electron density distributions in momentum space, i.e., electron momentum distribution (EMD).[1–8] The plane-wave impulse approximation (PWIA) calculations generally reproduce the measured EMDs for electron momentum below 1.5 a.u. (atomic unit) but tends to underestimate the experimental results in the large momentum region. For the atomic targets, the distorted-wave impulse approximation (DWIA) and distorted-wave Born approximation (DWBA)[9,10] have effectively addressed the limitations of the PWIA in fully reproducing the experiments. However, for molecular systems, the discrepancies between experiments and calculations may arise from multifaceted factors. For some molecular orbitals, the PWIA calculation fails to reproduce the intensity of the experimental EMDs near zero momentum.[11–16] Molecular vibrations were found to have noticeable contribution to the EMD’s intensity at low momentum region according to the harmonic analytical quantum mechanical (HAQM)[17] approach calculation, the classical molecular mechanics force field (MM3), and Born–Oppenheimer molecular dynamics (BOMD)[18] simulation, as well as the thermally sampling molecular dynamics (TSMD)[19,20] simulation. Moreover, the distorted wave was regarded as another physical factor that contributes to the high intensity at low momentum. This effect can be evaluated by experimentally varying the collision energy,[21–23] or by theoretically calculating the (e, 2e) triple differential cross sections (TDCSs) within the multicenter three-distorted-wave (MCTDW) method developed recently by Gong et al.[21–23]
Toluene (C7H8), one of the simplest substituted monocyclic aromatic compounds, is a volatile organic compound commonly found in urban pollution, primarily originating from fossil fuel combustion, industrial processes, and biomass burning.[24] Its contribution to photochemical smog and ozone formation in the troposphere underscores its environmental significance.[25] Moreover, due to its carcinogenic and mutagenic properties, elevated levels of toluene in human breath have been associated with lung cancer and smoking,[26,27] highlighting the importance of investigating this compound. The electronic structure of toluene has been studied using photoelectron spectroscopy with He I and He II radiation sources,[28–33] in which the ionization energies of valence orbitals were presented. Recently, Yamazaki et al. reported an EMS study on the toluene S1 state.[34] To our best knowledge, no comprehensive EMS study on the ground-state toluene molecule has been reported.
In this work, the valence electronic structure of toluene is investigated by EMS both experimentally and theoretically. The EMDs of valence orbitals were measured using a high-sensitivity electron momentum spectrometer. Theoretical calculations taking molecular vibrations into account through HAQM and TSMD approaches were performed within the PWIA. Moreover, the TDCSs were also calculated by the MCTDW method. The effects of molecular vibrations and the distorted waves on the valence orbital EMDs of toluene are analyzed by the comprehensive comparison between experimental measurements and theoretical predictions.
-
EMS is based on the binary (e, 2e) process, i.e., fast electron-impact single ionization,
Here, the subscripts i, s, and e refer to the incident, scattered, and ejected electrons, respectively, with corresponding energies E and momenta p. T represents the target atom or molecule, and T+ denotes the residual ion.
For the binary (e, 2e), the collision happens between the incident and the orbital electrons, and the residual ion can be regarded as a spectator. According to the conservation of energy and momentum, the binding energy ε and momentum p of the orbital electron can be derived by
The present EMS experiment was carried out on a high-sensitivity electron momentum spectrometer employing non-coplanar symmetric geometry.[35] Here, a brief description on the experiment is presented. For the details, please refer to Ref. [35]. In the experiment, the gaseous toluene molecule is ionized by 800-eV electron collision. The scattered and ejected electrons, with the equal energies (Es = Ee) and symmetric emission polar angles (θs = θe = 45°), pass through the conical retarding lenses and a double toroidal electrostatic analyzer and are detected by a two-dimensional (2D) position sensitive detector (PSD) positioned the exit of the analyzer. The PSD records the arrival times and positions of the two outgoing electrons, the former ensuring time coincidence of them and the latter enabling the determination of their momenta and energies. Therefore, the orbital electron binding energy can be obtained according to Eq. (2). The magnitude of the electron momentum in Eq. (3) can be given by
where ϕ is the relative azimuthal angle between the scattered and ejected electrons.
Prior to the toluene experiment, the instrumental energy resolution (∼ 3.0 eV at FWHM) and angular resolution (Δθ = 1.2°, Δϕ = 2.2°) were determined through the experiment on Ar 3p ionization.
On the theoretical side, within the framework of the MCTDW[21,22] method, the TDCS for randomly oriented molecular target can be expressed as
Here, the ionization amplitude Tfi is given by
In this equation,
If taking the place of the distorted waves by the plane waves, the MCTDW can reduce to the PWIA, expressed as
For molecules, within the Born–Oppenheimer approximation, neglecting molecular rotation and applying the closure relation to all final vibrational states, the TDCS can be further simplified as[17]
where M(p) is the structure factor, xiv(Q) is the vibrational wave function in initial state, v is the vibrational quantum number, Q represents the molecular geometry, and pv(T) is the population at temperature T. ρ(p;Q) is the spherically averaged momentum density distribution, which can be written as
where Sα(Q) represents the spectroscopic factor (also known as pole strength), which quantifies the likelihood of a single-hole configuration in the resulting ionic state.
To investigate the influence of molecular vibrations on EMDs, Watanabe et al.[17] proposed an HAQM approach, wherein the vibrational wave function xiv(Q) is expressed as a product of harmonic oscillator functions ξvL(QL). Here, QL represents the displacement along the normal coordinate of the L-th vibrational mode from the equilibrium geometry. By assuming that ρ(p;Q) varies slowly with respect to Q near equilibrium and neglecting high-order cross-terms between distinct normal modes, the structure factor M(p) can thus be written as
Here, the first term ρ(p;Q0) represents the EMD at the equilibrium geometry Q0, and the second term corresponds to the vibrational effects from all vibrational normal modes. qL is the unit vector along the normal coordinate.
The TSMD sampling[19,20] is another approach, in which both harmonic and anharmonic vibrations are taken into account. The numerical solution of Eq. (8) can be expressed as
Molecular geometries are sampled at a specific temperature according to the vibrational population
When N is large enough, a full Q-space sampling can be achieved.
In this work, the equilibrium geometry of toluene is optimized using density functional theory (DFT) with the B3LYP hybrid functional[36,37] and Dunning’s aug-cc-pVTZ basis set.[38] The position-space wave functions, vibrational frequencies, and normal coordinates are computed at the B3LYP/aug-cc-pVTZ level. The function ρ(p;Q0 + QLqL) is calculated using the B3LYP/aug-cc-pVTZ and fitted employing a fourth-order polynomial function, based on the displacement QL chosen from |ξvL(QL)|2/|ξvL(0)|2 = 3/4, 1/2, 1/4, 1/8, 1/16. The theoretical EMDs considering the vibrational effects of each mode with Boltzmann distributions at room temperature (298 K) are calculated according to Eq. (10). The TSMD simulation is performed using a thermostat at a temperature of 298 K to simulate the vibration of the neutral toluene molecule. The molecular geometry is taken every 2.5 fs after a 5-ps equilibration run, and a total of 2000 geometries are sampled using Wigner distributions at T = 298 K via the Newton-X package.[39] The final EMD is obtained as the average of these configurations according to Eq. (12). All the calculations are performed using the Gaussian 09 program,[40] except for the MCTDW whose codes were developed by Gong et al.[21]
-
The molecular point group of toluene is Cs, and its ground-state electronic configuration calculated by B3LYP/aug-cc-pVTZ is as follows:
The valence molecular orbitals (MOs) of toluene consist of four inner-valence orbitals and fourteen outer-valence orbitals. The experimental 2D electron density map (2D map) for the valence MOs of toluene is displayed in Fig. 1(a). From this map, information on the orbital symmetry, electron binding energy, and momentum distribution can be extracted.
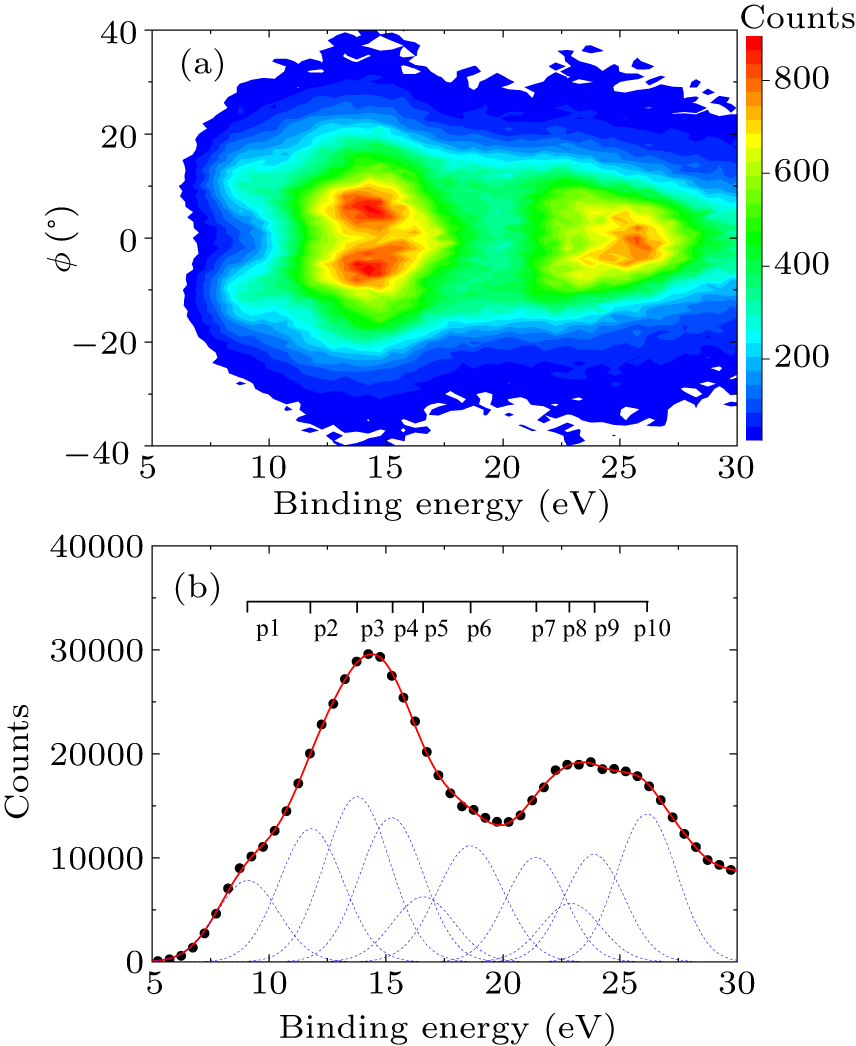
The experimental binding energy spectrum (BES) of toluene was obtained by integrating the 2D map over the relative azimuthal angles, as depicted in Fig. 1(b). The BES spans an energy range from 5 eV to 30 eV which covers the whole valence orbitals. Due to the limit of our instrumental energy resolution, only a “shoulder” at around 9 eV and two broad peaks at about 14 eV and 24 eV are observed clearly in the BES, which is similar to the BES presented by Yamazaki et al.[34] The previous high-resolution PES[28] showed six separated peaks corresponding to fourteen outer-valence MOs and four weak peak structures representing four inner-valence orbitals.
To extract the EMDs, a series of binding energy spectra for different ϕ angles are derived from the measured 2D map. Ten Gaussian peaks (labeled p1–p10) are used to deconvolute these spectra, respectively. The peak positions of these Gaussian functions are referred to the PES results,[28,29,31] while the peak widths are the combination of our instrumental energy resolution and the Franck–Condon envelopes derived from the PES.[28,31] In the outer-valence region, the peak p1 at ∼9.1 eV (“shoulder”) corresponds to the highest occupied molecular orbital (HOMO) 11a′ and the next HOMO 7a″. Peak p2 at ∼ 11.8 eV represents the next three orbitals (6a″, 10a′, 9a′) while p3 at ∼ 13.8 eV stands for the four orbitals (5a″, 8a′, 4a″, 7a′). The following three Gaussian peaks (p4–p6) at ∼ 15.2 eV, ∼ 16.6 eV, and ∼ 18.6 eV correspond to MO sets (6a′, 3a″), (5a′), and (4a′, 2a″), respectively. In the inner-valence region above 20 eV, the last four Gaussian peaks (p7–p10) at ∼ 21.4 eV, ∼ 22.9 eV, ∼ 23.9 eV, and 26.1 eV correspond to 3a′, 1a″, 2a′, and 1a′ orbitals, respectively. The information on the positions of these Gaussian peaks (p1–p10) and the corresponding valence orbital ionization energies of toluene molecule is summarized in Table 1, along with the measured values from the previous PES experiments[29,31] for comparisons.
The experimental EMDs for p1–p10 were obtained by plotting the areas of Gaussian peaks derived from the deconvolution of the BES relative to different ϕ (i.e., momentum), as depicted in Figs. 2–8, where error bars of experimental data included the deconvolution and statistical uncertainties. The theoretical EMDs, calculated by the PWIA at equilibrium geometry (PWIA), the PWIA considering vibrations through HAQM (PWIA/Vib) or TSMD sampling (TSMD), as well as the MCTDW methods, are also shown for comparisons. To facilitate comparison with the experimental EMDs, the instrumental angular resolution (Δθ = 1.2°, Δϕ = 2.2°) was folded into the theoretical EMDs using the Gaussian-weighted planar grid method.[41] Furthermore, normalization between the theoretical and experimental EMDs for all the valence orbitals was carried out by using a common factor. This factor is determined by putting the experimental EMD for p1 and the theoretical EMD of the HOMO 11a′ and next HOMO 7a″ on the same scale.
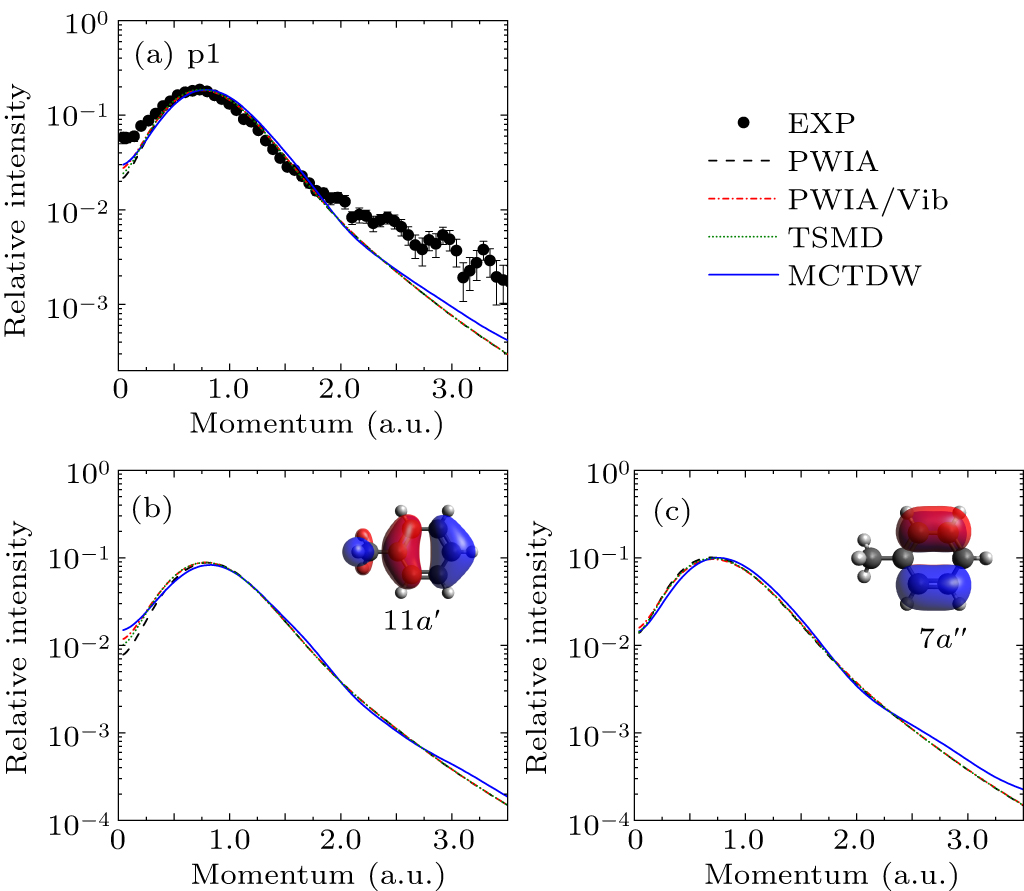
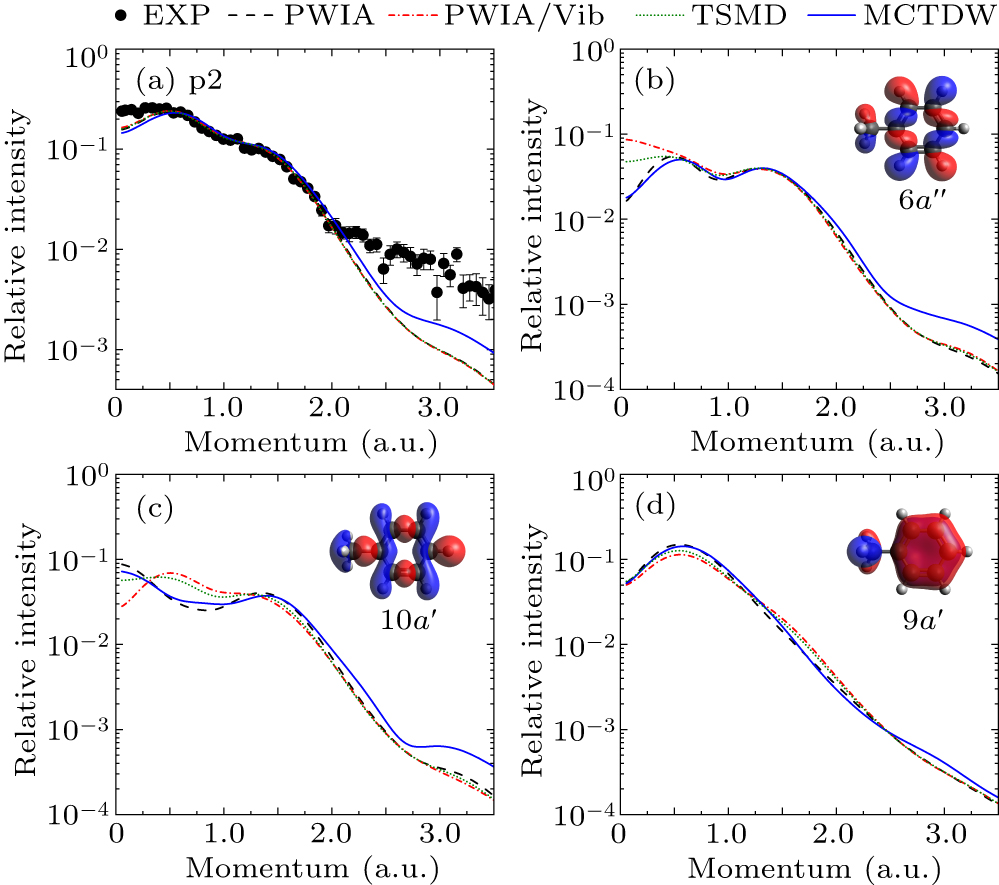

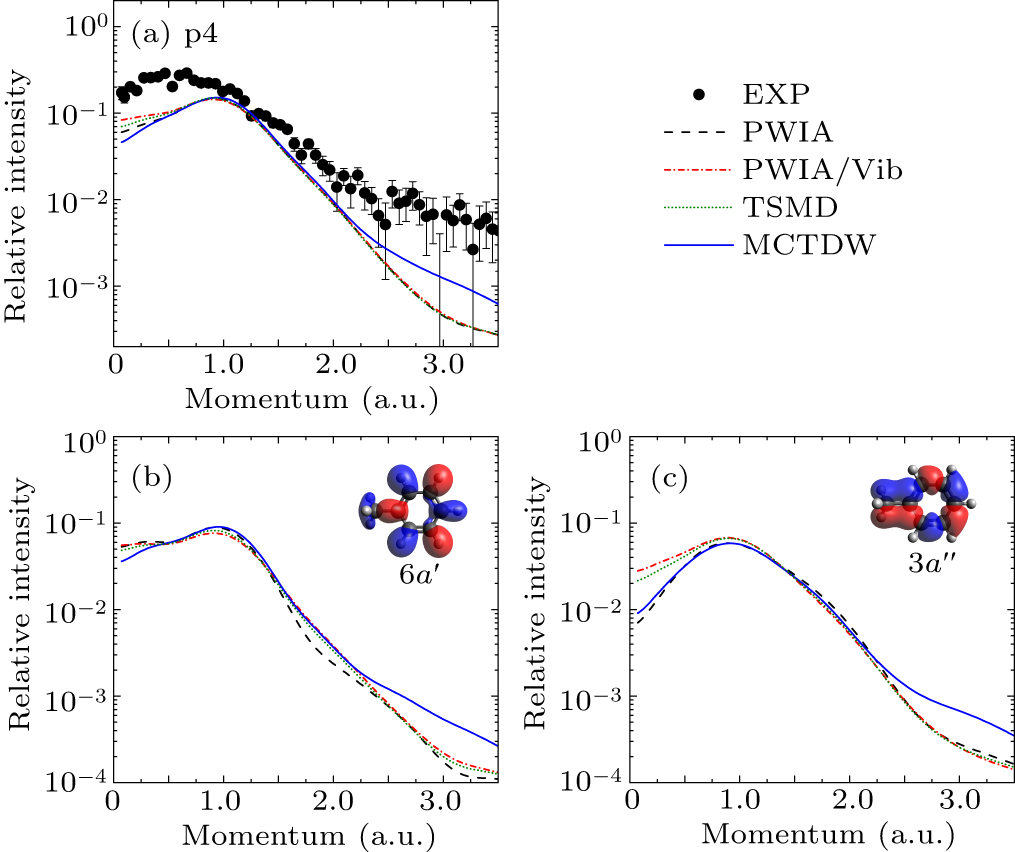
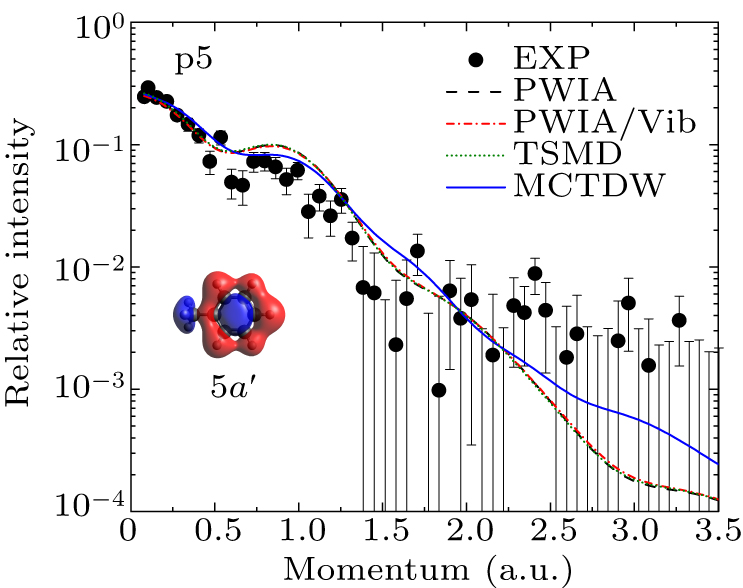
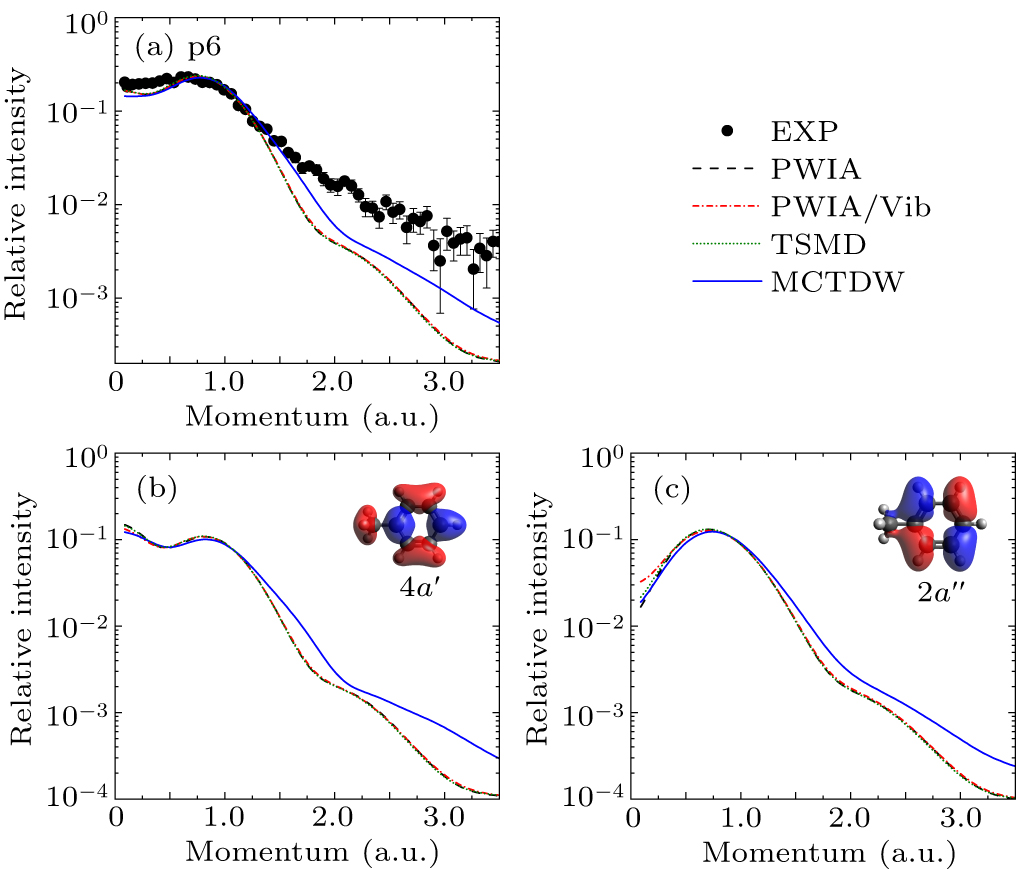
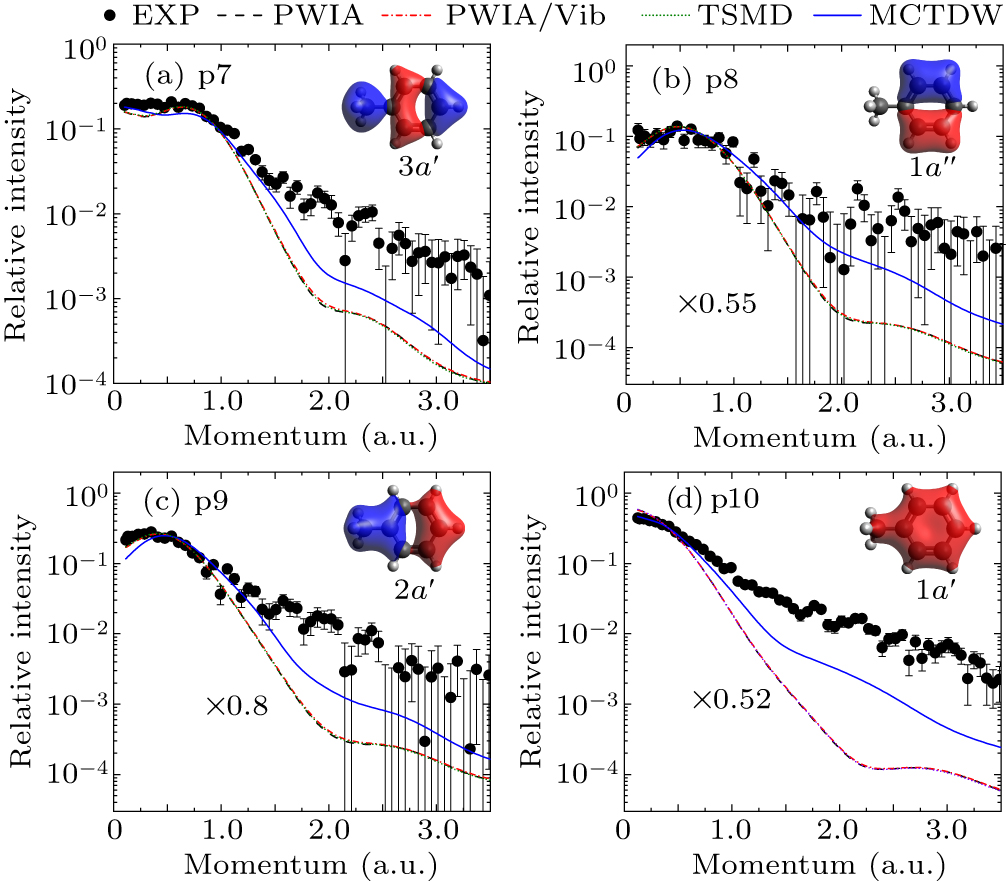
Figure 2(a) presents the experimental and theoretical EMDs for p1, corresponding to the joint contributions of the HOMO 11a′ and next HOMO 7a″. Both of them exhibit a typical p-type character with a maximum at p ∼ 0.75 a.u. All theoretical calculations underestimate the experimental results in the low or high momentum region. The calculations considering molecular vibrations by the PWIA/Vib and TSMD slightly increase the intensity at small momentum relative to the PWIA result, whereas the MCTDW calculations have an obvious improvement both in small and large momenta, suggesting the result of the distorted-wave effect. The theoretical EMDs for the individual MO, 11a′ and 7a″, are shown in Figs. 2(b) and 2(c). For 11a′ orbital, compared to the PWIA calculation, the PWIA/Vib, TSMD, and MCTDW calculations predict higher intensities in the low momentum region, showing the visible effects of molecular vibrations and distorted waves. While for 7a″ orbital, all the calculations present the similar results in the low momentum region. This difference may be ascribed to their distinct orbital characters, as the orbital maps showed in Figs. 2(b) and 2(c).
Figure 3 displays the experimental EMD for p2 and the corresponding theoretical calculations for orbitals 6a″, 10a′, and 9a′. The orbital maps show that these orbitals are all composed of C–C and C–H bonds, but they have obviously different symmetries. This diversity makes their EMDs distinct, as shown in Figs. 3(b)–3(d). The calculations show that the vibrational effects on the EMDs are observed in the low momentum region while the distorted-wave effects appear in the high momentum region. Specifically, the PWIA calculations reveal that the EMD of 6a″ exhibits a double-peak structure with two maxima, while that of 10a′ exhibits a typical sp-type character. However, when the vibrational effects are considered in the PWIA/Vib. and TSMD calculations, the behaves of the EMDs of these two MOs are reversed. This phenomenon arises from Herzberg–Teller vibronic coupling, which results in orbital mixing. For MO 9a′, the vibrational effects slightly change the intensity of the EMD. As for the distorted-wave effect, it primarily presents for 6a″ and 10a′, as indicated by the MCTDW calculations, while almost invisible for 9a′. However, for the summed EMDs of these orbitals as displayed in Fig. 3(a), the calculated results show no obvious variation except for the MCTDW. All calculations are generally consistent with the experimental EMD.
As mentioned above, the peak (p3) at ∼ 13.8 eV in the BES is owed to the joint contribution of 5a″, 8a′, 4a″, and 7a′ orbitals. The experimental and theoretical EMDs for p3 are shown in Fig. 4(a). The PWIA calculations without or with molecular vibrations involved (i.e., PWIA, PWIA/Vib, and TSMD) present almost the same EMDs, whereas the MCTDW calculation enhance the EMD’s intensity at large momentum region. In general, all the calculations are consistent with the experimental EMD in the low-momentum region. For the individual MO, as shown in Figs. 4(b)–4(e), 5a″ and 8a′ orbitals are contributed from the C–H and C–C bonds of methyl and benzene. The 4a″ orbital only involves the C–H and C–C bonds of benzene while 7a′ orbital mainly comes from the methyl and its adjacent carbon atoms. The PWIA/Vib and TSMD calculations show that molecular vibrations have obvious contribution to the EMDs for these four orbitals in the low momentum region. Owing to their nearly degenerate energies, these four orbitals undergo Herzberg–Teller vibronic coupling, which leads to mutual borrowing the intensity of their EMDs. As a result, although the individual orbital EMDs calculated by PWIA, PWIA/Vib, and TSMD are different, the summed EMDs for these four orbitals remain remarkably similar. The MCTDW calculations suggest that the distorted-wave effects on the EMDs appear in large momentum region, improving the agreement between the calculated and measured EMDs as shown in Fig. 4(a).
As shown in Fig. 5, the 6a′ orbital of toluene is dominated by C–H bonds while the 3a″ mainly consists of C–C bonds. The introduction of methyl breaks the orbital symmetry of the benzene ring, which leads to the nonzero intensity of their EMDs near the origin of momenta. The PWIA/Vib and TSMD calculations indicate that molecular vibrations have a significant influence on the EMD of 3a″ orbital in low momentum region, but only a slight effect on the 6a′ orbital. The MCTDW calculations revealed the effects of the distorted waves on the EMDs of 6a′ and 3a″ in large momentum region. Figure 5(a) shows the summed theoretical EMDs in comparison with the experimental data. All the calculations predict the shape of the measured EMD but greatly underestimate the intensity. The PWIA/Vib and TSMD calculations improve the intensity at low momenta while the MCTDW calculations enhance the intensity at large momenta. However, the discrepancy between the calculated and measured EMDs remains, suggesting the limitations of the present theoretical approaches.
Figure 6 shows the experimental and theoretical EMDs for p5, which corresponds to the 5a′ orbital of toluene, comprising contributions from both the benzene ring and the methyl group. Both the experimental and calculated EMDs exhibit a typical sp-type character. The MCTDW calculation reproduces the experiment better than the other three calculations. This suggests that the distorted-wave effect is noticeable for this orbital. The PWIA, PWIA/Vib, and TSMD calculations yield nearly identical EMDs, suggesting no apparent vibrational effect.
The experimental and theoretical EMDs for p6 are displayed in Fig. 7(a). This peak contains two MOs shown in Figs. 7(b) and 7(c). The 4a′ orbital shows a symmetric component of the benzene ring along with methyl contribution, while the 2a″ orbital is dominated by an antisymmetric component of benzene illustrating a pseudo-π orbital character. These differences between 4a′ and 2a″ orbitals lead to distinct EMDs, i.e., sp-type EMD for 4a′ orbital and p-type for 2a″ orbital. The calculated EMDs indicate that molecular vibrations have negligible effects on the 4a′ orbital and minor effects on the 2a″ orbital, whereas distorted-wave effects are significant for both. Compared with the experiment, all theoretical calculations can reproduce the shape of the experimental EMD, but underestimate the intensity at momenta above 1.5 a.u. The results calculated by PWIA, PWIA/Vib, and TSMD indicate no observable vibrational effects. In contrast, the MCTDW calculation reduces the discrepancy between the calculated and measured EMDs, which is the result of the distorted-wave effect.
Figures 8(a)–8(d) present the experimental and theoretical EMDs of the inner-valence MOs 3a′, 1a″, 2a′, and 1a′, respectively. For these four orbitals, the PWIA, PWIA/Vib, and TSMD calculations give the same EMDs, indicating the absence of molecular vibrational effects. In contrast, the MCTDW calculation greatly enhances the EMD’s intensity in large momentum region, revealing the significance of the distorted-wave effect for these inner-valence orbitals. Compared with the measurement, for the 3a′ orbital, the MCTDW calculation depicts the experiment better than the other three calculations. For 1a″, 2a′, and 1a′ orbitals, additional factors are needed when comparing the calculated EMDs with the measured ones, which represent the pole strength of ionizations from these orbitals. As shown in Figs. 8(b)–8(d), the calculated EMDs are multiplied by factors of 0.55 for 1a″, 0.8 for 2a′, and 0.52 for 1a′, respectively, to enable comparison with the experimental data. Obviously, the MCTDW calculation provides better agreement than the others.
-
The EMDs of valence orbitals of toluene were obtained experimentally using an electron momentum spectrometer. Through the comparison between experimental EMDs and theoretical calculations (PWIA, PWIA/Vib., TSMD, and MCTDW), it was found that molecular vibrational effects are evident for the outer-valence orbitals involving significant methyl contributions but absent for the inner-valence orbitals, while the distorted-wave effects are pronounced in high momentum region for all valence orbitals and also in low momentum region particularly for the 5a′ and 3a′ orbitals. The present results provide a deep insight into the valence electronic structures of toluene molecule.
 DownLoad:
DownLoad: