
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
钙钛矿或钙钛矿结构化合物中存在自旋-自旋和自旋-轨道间的相互作用,展示出丰富的物理行为,引发了大量关注。钙钛矿是一种常见的晶体结构,普遍的表达式为ABX3(ABX3型物质不一定都形成钙钛矿),其中:A位是半径较大的阳离子,如碱土金属或者稀土元素离子,配位数为9~12;B位是半径较小的阳离子,如过渡族金属离子,配位数为6;X位是半径较大的阴离子,如氧、氟离子等。B和X离子构成BX6八面体,相邻八面体之间以顶点的X离子连接,形成三维结构。理想情况下,钙钛矿结晶为立方晶系(空间群:Pm
$ \overline 3 $ m),图1(a)显示了其晶体结构。实际材料中,立方钙钛矿可以畸变为其他形式。Goldschmidt[1]引入容忍因子t,用于定义晶体结构偏离立方钙钛矿的畸变程度
式中:rA、rB和rX分别为A、B和X离子的半径。对于钙钛矿材料,分为3种情况:(1) 当0.9 < t ≤1.0时,材料为立方结构;(2) 当0.8 < t ≤ 0.9时,晶格发生扭曲畸变,BX6八面体绕某一方向旋转,材料为低对称性结构;(3) 当t > 1.0时,材料可能形成六方、三方或单斜等层状结构。
第1种情况对应的是未发生畸变的立方钙钛矿,如图1(a)所示。在第2种情况下,全部BX6八面体通过顶点的X离子连接,与立方钙钛矿中的连接方式一致。BX6八面体具有多种旋转方式[2],可以形成不同类型的畸变钙钛矿,如四方、正交或单斜结构。空间群为Pnma的正交钙钛矿是一种常见的畸变钙钛矿,其晶体结构如图1(b)所示。
对于第3种情况,A离子与X离子半径接近,它们在垂直于c轴的平面上以1∶3的比例密堆排列,B离子填充在近邻平面所形成的八面体空隙中,部分或全部BX6八面体以X3平面连接,形成六方(如2H、4H、6H)、三方(如3R、9R、12R)或单斜(如9M、5M、6M)等层状结构。这类含有BX6八面体共面连接的ABX3型化合物一般称为六方钙钛矿。晶体结构可采用数字组合字母的方式表示,其中,数字表示一个晶胞内含有AX3平面的个数(三方结构采用六方晶胞时,AX3平面个数是实际的3倍;单斜结构的数字根据与之相似的六方或三方结构而来),字母表示结构类型(H、R和M分别代表六方、三方和单斜结构)。当BX6八面体共面连接时,A的配位数仍为12,但配体X的排列方式与八面体共顶点连接时的情况不同,如图2所示。对于第1和第2种情况的钙钛矿,立方、四方、正交和单斜等结构均表示为3C。
在高温高压环境下,当A离子半径较大时,ABX3易形成不同类型的六方钙钛矿。六方钙钛矿中多个BX6八面体共面连接可形成B2X9、B3X12、B4X15等多聚体。在多聚体内部,相邻的B离子距离较近,B离子之间的相互作用比钙钛矿更复杂,从而呈现出一些与钙钛矿不同的物理性质。在足够高的合成压力下或B离子半径增大时,六方钙钛矿可以转变为钙钛矿。下文中ABX3型化合物的多层堆积变体包括六方钙钛矿和钙钛矿。
Ba2+具有较大的半径[3],可以与O2−形成密堆积。当B为过渡金属离子M时,BaMO3易形成六方钙钛矿。当改变合成条件时,它们可以形成不同类型的六方钙钛矿,也可能形成钙钛矿。六方钙钛矿中,共面连接八面体的存在可能会对这类化合物的电学和磁学性能产生巨大影响,是材料物理学中许多量子材料定向研究的主题[4]。因其潜在的低磁矩或轨道占有率排序的几何挫折,BaMO3六方钙钛矿是低温下量子自旋液体的候选者,在低温下可能存在量子临界点。在BaMO3中,随着合成压力的增加,2H→9R→4H→6H→3C是常见的演化顺序,MO6八面体共顶点连接的比例逐渐增加,由于共顶点连接的八面体中M离子的间距大于多聚体内M离子的间距,上述转变被认为是M离子在压力下重新分布以减少静电排斥。此外,对于同一M,BaMO3钙钛矿可能表现出与六方钙钛矿不同的物理性质,如BaRuO3钙钛矿具有与六方钙钛矿不同的巡游铁磁性。因此,本文主要讨论BaMO3多层堆积变体所展示的复杂晶体结构和物理性质,并探索结构和物性随M离子半径及合成压力变化的演化规律。
-
常压下合成的BaTiO3为六方钙钛矿[5],属于六方晶系,空间群为P63/mmc。由于一个晶胞内沿c轴方向有6个BaO3层,该结构表示为6H相,晶体结构见图3(a)。在ABX3材料的多层堆积变体中,分别用c和h代表立方和六方堆积,其中,c表示上下2层BaO3平面的原子排列方式不同(见图2(a)),h表示上下2层BaO3平面的原子排列方式相同(见图2(b))。在6H相中,MO6八面体和M2O9二聚体共顶点连接,沿c轴方向交替排列,所以BaO3层的堆积序列为(hcc)2。图3(b)显示了BaTiO3的温度-压力(T-p)相图[6],在不同的条件下,BaTiO3可以形成多种钙钛矿,包括立方、四方、三方、正交相等。例如,在3.2 GPa的压力下,BaTiO3在298 K即可转变为立方钙钛矿。
-
在常压下合成的BaVO3为5H结构[7],属于三方晶系,空间群为P
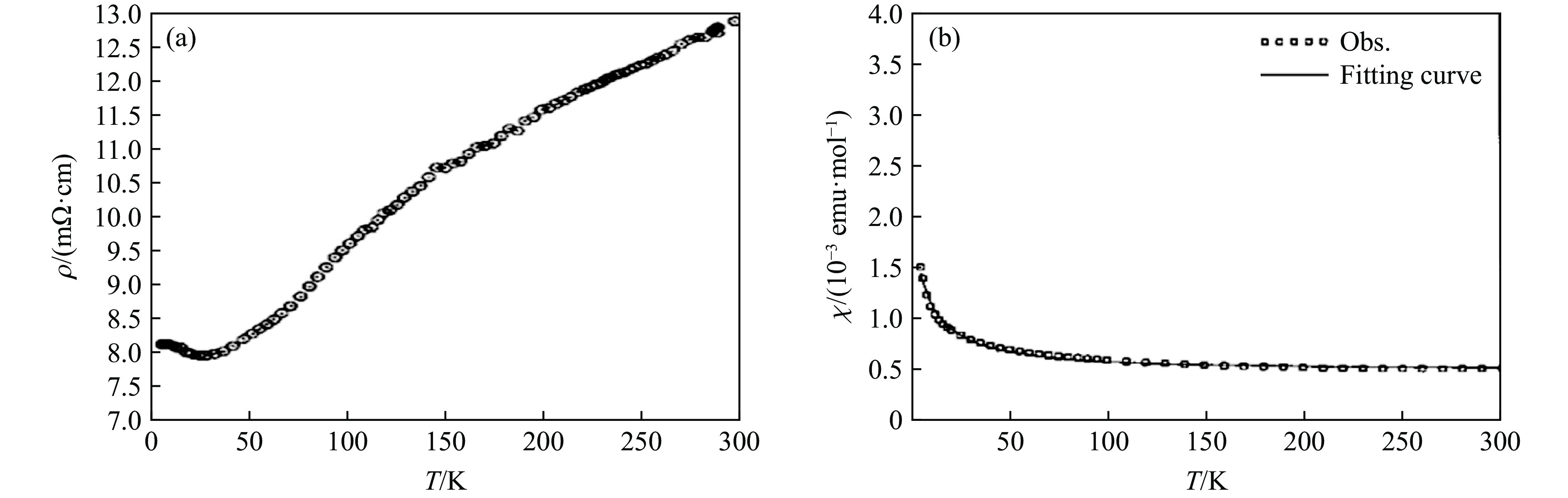
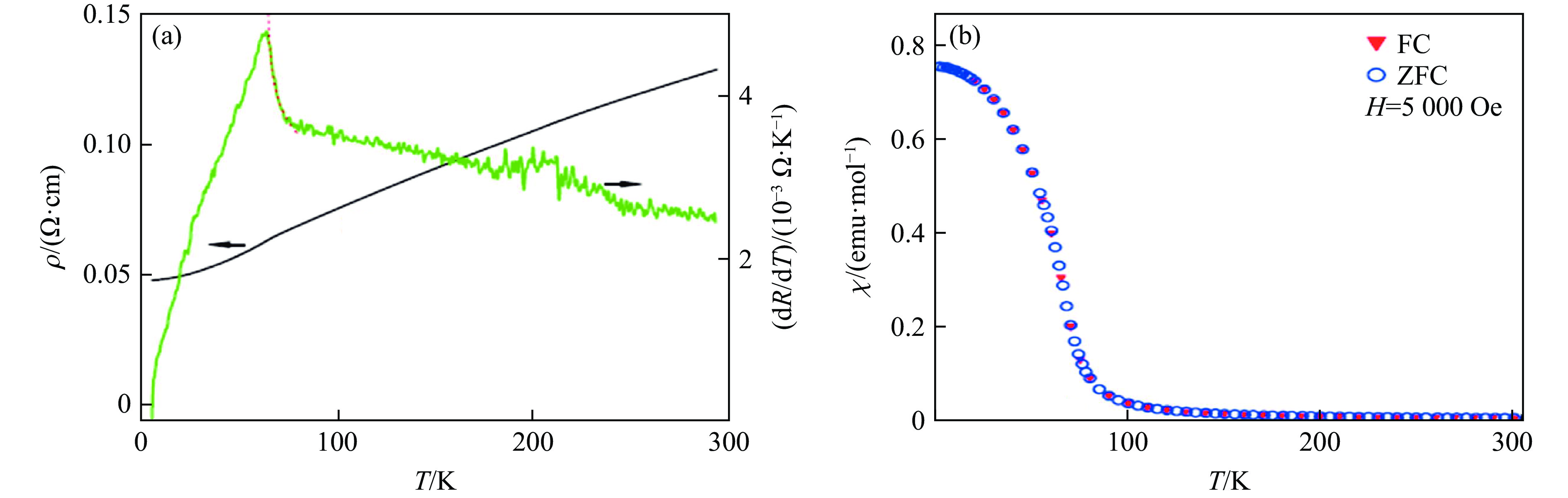
$ \overline 3 $ m1。BaVO3在15 GPa、1350 ℃的合成条件下形成立方钙钛矿(见图1(a))[8]。利用复杂的化学方法或者高温高压方法(6.0~6.5 GPa、1200 ℃),BaVO3可以形成14H相(六方结构,空间群P63/mmc)[9]。5H和14H相的晶体结构如图4所示。在5H相中,1个M3O12三聚体与2个MO6八面体共顶点连接,沿c轴交替排列;在14H相中,3个M2O9二聚体共顶点连接,然后与1个MO6八面体共顶点连接,沿c轴交替排列。因此,在5H和14H相中,BaO3平面的堆积序列分别为chhcc和(hchchcc)2。如图5[7]所示,5H-BaVO3的电阻率-温度(ρ-T)曲线在25 K附近存在转折点,即发生了金属-半导体转变。5H-BaVO3具有顺磁性,其低温磁化率-温度(χ-T)曲线满足
式中:C、θ和χ0分别为居里常数、外斯温度和与温度无关的顺磁磁化率。通过对χ-T曲线进行拟合,得到χ0 = 0;顺磁有效磁矩μeff为0.305μB (μB为波尔磁子),比V4+的理论值1.73μB小得多;外斯温度θ为−6.9 K,说明V4+离子之间的反铁磁相互作用占主导地位。
图6表明3C-BaVO3是顺磁性金属[8],电阻率较大。低温时,ρ与T的关系满足
式中:ρ0为剩余电阻率,A为特征常数,n为电阻指数。由图6可得:ρ0 = 4.07 Ω∙cm,n = 2,说明3C-BaVO3属于费米液体金属。
-
图7显示了BaCrO3的压力-温度相图[10]。在高温高压合成条件下,BaCrO3形成了5H(图4(a))、4H、6H(图3(a))和3C(图1(a))等多层堆积变体[10–12]。4H相属于六方晶系,空间群为P63/mmc,图8(a)显示了它的晶体结构,其中,2个MO6八面体共面连接,形成M2O9二聚体,相邻的M2O9二聚体共顶点连接,沿c轴方向交替排列,因此,BaO3层的堆积序列为(hc)2。
当压强在6.0~6.5 GPa区间,合成温度在
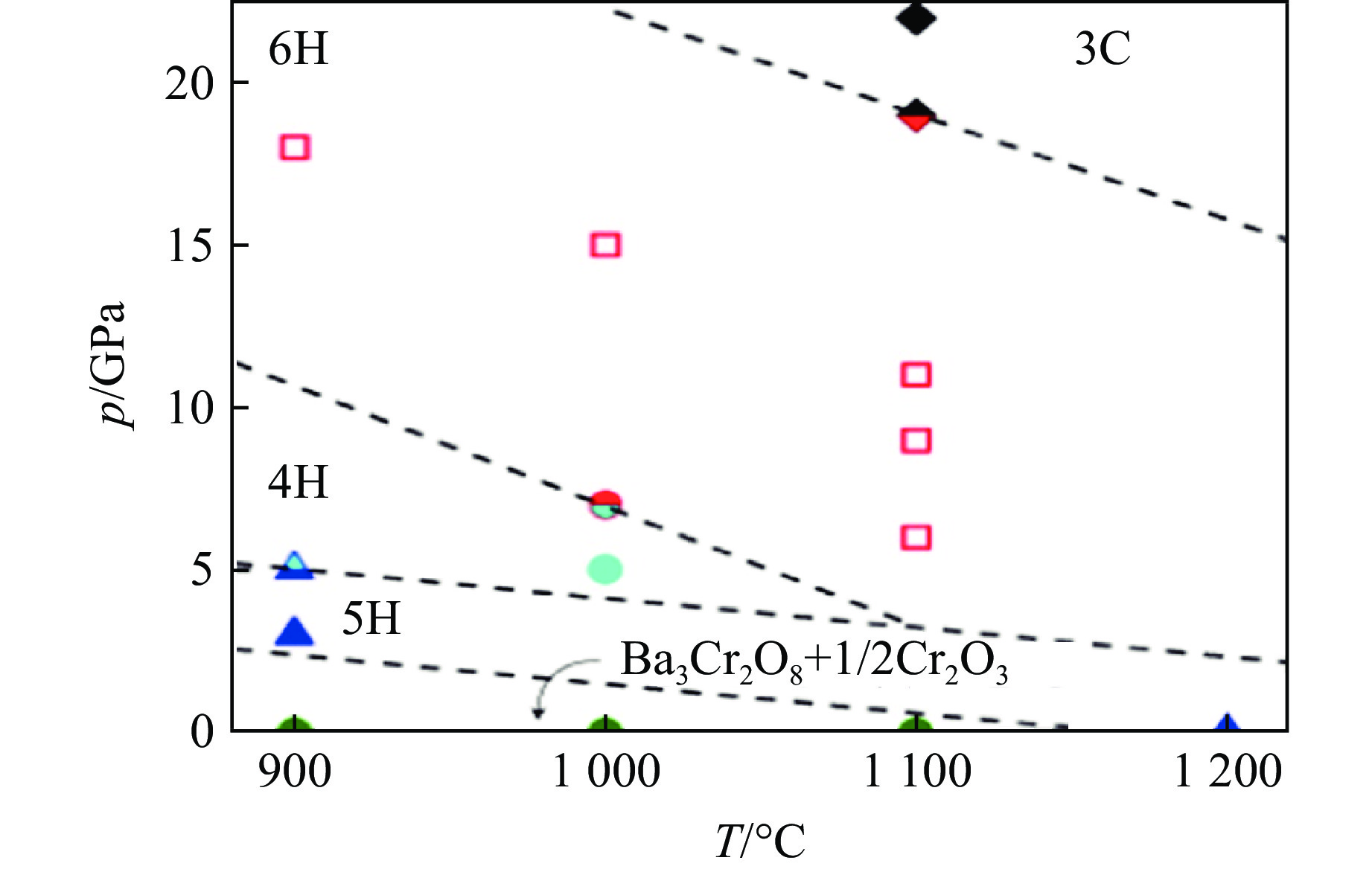
1200 ~1300 ℃区间时,BaCrO3可形成14H(图4(b))、12R、27R等多层堆积变体[13–15]。图8(b)和图8(c)分别显示了12R和27R的晶体结构。在12R相(三方结构,空间群为R$ \overline 3 $ m)中,1个M3O12三聚体与1个MO6八面体共顶点连接,沿c轴交替排列;在27R相(三方结构,空间群为R$ \overline 3 $ m)中,4个M2O9二聚体共顶点连接,然后与1个MO6八面体共顶点连接,沿c轴交替排列。因此,在12R和27R相中,BaO3平面的堆积序列分别为(hhcc)3和(hchchchcc)3。Cr离子的巡游性较差,所以BaCrO3为电阻率很大的绝缘体,文献[10]对BaCrO3物理性质的研究主要集中在磁学性质方面。如图9所示,5H-BaCrO2.8具有反铁磁性,其奈尔温度TN为250 K[10],48 K下零场冷(zero field cooling, ZFC)和场冷(field cooling, FC)曲线的分离是由样品中的BaCr2O4杂相所致。利用式(2)对260~300 K之间的数据进行拟合,得到μeff为3.4μB,介于Cr4+的理论值2.83μB和Cr3+的理论值3.87μB之间;θ为−684 K,说明Cr离子之间的反铁磁自旋相互作用占主导地位。
图10(a)显示6H-BaCrO3具有亚铁磁性,其居里温度TC为192 K[16],利用式(2)对275~300 K之间的数据进行拟合,得到μeff为2.51μB,接近Cr4+的理论值2.83μB;θ为−316 K,说明Cr离子之间的反铁磁相互作用占主导地位。图10(b)中,2 K下的磁滞回线说明6H-BaCrO3在低温下的每个Cr4+的饱和磁矩为0.09μB,与低温中子衍射结果一致。图10(c)显示了6H-BaCrO3在低温下的磁结构,Cr2O9二聚体中Cr离子的磁矩平行排列,CrO6八面体与Cr2O9二聚体中的Cr离子通过O离子连接,其中,Cr离子的磁矩反平行排列,所以,6H-BaCrO3整体上显示出亚铁磁性。
-
在常压、
1100 ℃的合成条件下,BaMnO3形成六方层状结构(2H相,空间群为P63/mmc)[17–18],图11(a)显示了2H-BaMnO3的晶体结构,在c轴方向上展现了2个晶胞,其中,八面体全部共面连接,沿着c轴方向呈链状排布。在高温高压合成条件下,BaMnO3形成了9R、4H(图8(a))和6H(图3(a))等六方钙钛矿结构[18–21]。在其他的常压高温合成条件下,BaMnO3形成了6H'(与6H相不同的另外一种六层结构)、8H、10H、15R、21R、27R'(与27R相不同)和33R等六方钙钛矿结构[22–28]。图11(b)~图11(g)显示了9R、6H'、8H、10H、15R和21R相的晶体结构。在6H'相(六方结构,空间群为P
$ \overline{6} $ m2)中,1个M2O9二聚体与1个M4O15四聚体共顶点连接,沿c轴交替排列;在8H相(六方结构,空间群为P63/mmc)中,相邻的M4O15四聚体共顶点连接,沿c轴交替排列;在10H相(六方结构,空间群为P63/mmc)中,M2O9二聚体和M3O12三聚体共顶点连接,沿c轴交替排列。9R、15R、21R、27R'和33R相均为三方结构,所属空间群为R$ \overline{3} $ m,结构中分别形成M3O12三聚体、M5O18五聚体、M7O24七聚体、M9O30九聚体和M11O36十一聚体,这些多聚体共顶点连接,沿c轴交替排列。因此,在6H'、8H、10H、9R、15R、21R、27R'和33R相中,BaO3平面的堆积序列分别为hchhhc、(hhhc)2、(hhchc)2、(hhc)3、(hhhhc)3、(hhhhhhc)3、(hhhhhhhhc)3和(hhhhhhhhhhc)3。与BaCrO3类似,BaMnO3也是电阻率很大的绝缘体。图12(a)显示了2H-BaMnO3的χ-T曲线[17]。2H-BaMnO3在低温下表现出反铁磁性,每一个链中近邻自旋均为反平行排列,其TN = 59 K,在1.7 K时,每个Mn4+的磁矩为1.31μB。图12(b)和图12(c)[22]显示,4H-BaMnO3和6H-BaMnO3在低温下均表现出反铁磁性,TN分别为263和220 K,6H相在低温下还出现一个反铁磁转变,低温相为倾斜反铁磁有序,对应的奈尔温度TN = 25 K。6H相的μeff为3.84μB,接近Mn4+的理论值3.87μB;θ为−219 K,说明Mn离子之间的反铁磁相互作用占主导地位。图12(d)显示了15R-BaMnO3的M-T曲线[26],其中,奈尔温度TN = 230 K。在短程有序温度Ts = 330 K下,15R-BaMnO3表现出短程磁有序。局域晶格畸变温度TD = 280 K对应于拉曼频率随温度的突变。文献[26–27]均未讨论15R相在40 K下的磁学性质。
-
在常压下合成的BaFeO2.5中,Fe-O多面体都以顶点上的O离子连接[29]。BaFeO2.5经大于600 ℃的高温氧化处理可得6H相(图3(a))[30–31]。BaFe[(CN)5NO]·3H2O经氧化热解可得10H相(图11(e))[32]。在高温高压合成条件下,BaFeO3可以形成12层结构(“12R”)[33],其空间群为R3m,如图13(a)所示。但最近的研究表明,BaFeO3在高温高压下应形成单斜畸变的12层结构(12M)[34],属于单斜晶系,空间群为C2/m,如图13(b)所示。图13(b)中,紫色边框为晶胞边界,绿色边框为从中划出12R结构的“晶胞边界”。在高温下(570 K),12M-BaFeO3形成12R结构(图8(b))[34]。BaFeO2.5在较低温度(200 ℃)下经氧化处理可得3C相(图1(a))[35–37]。
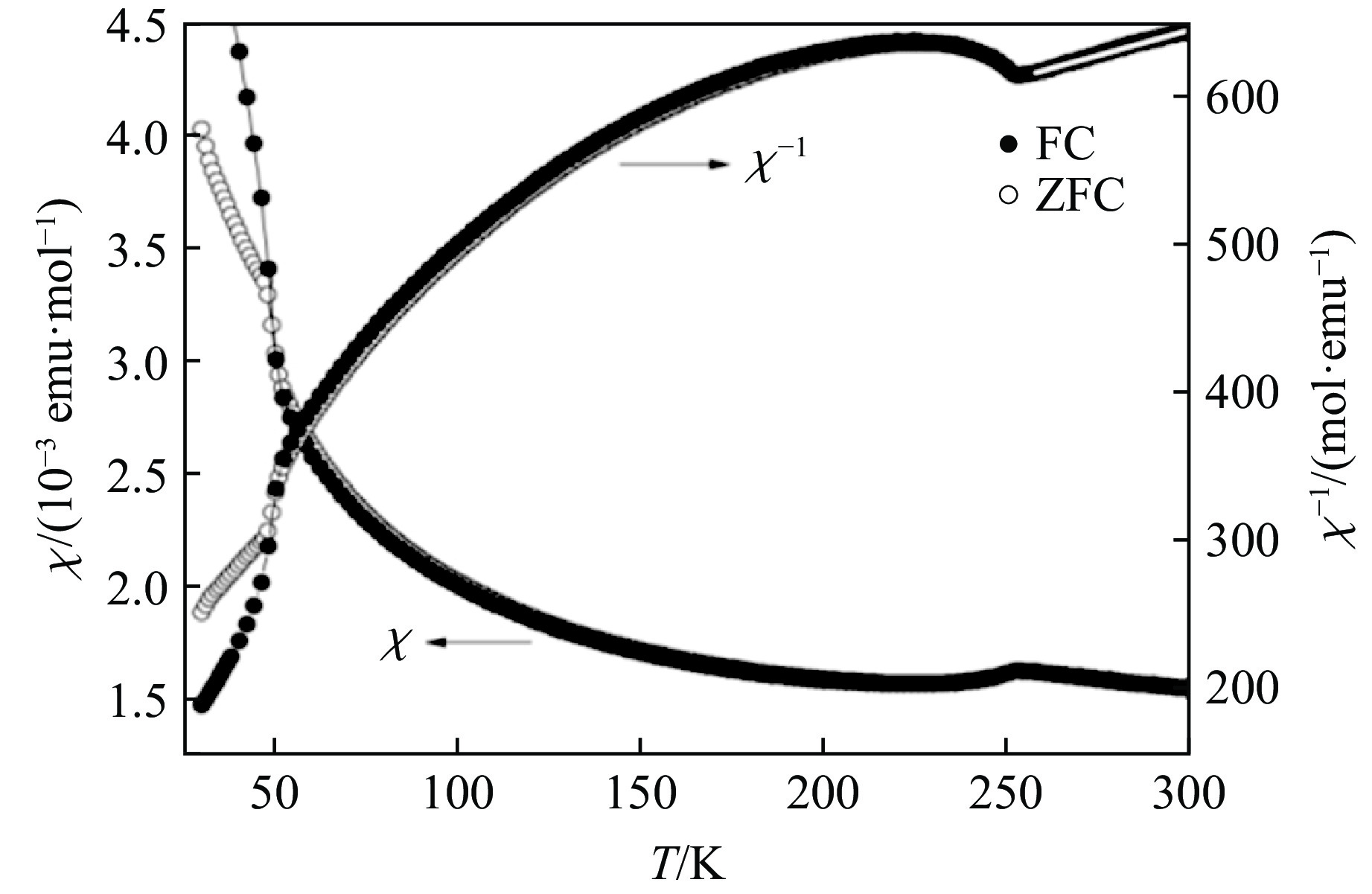
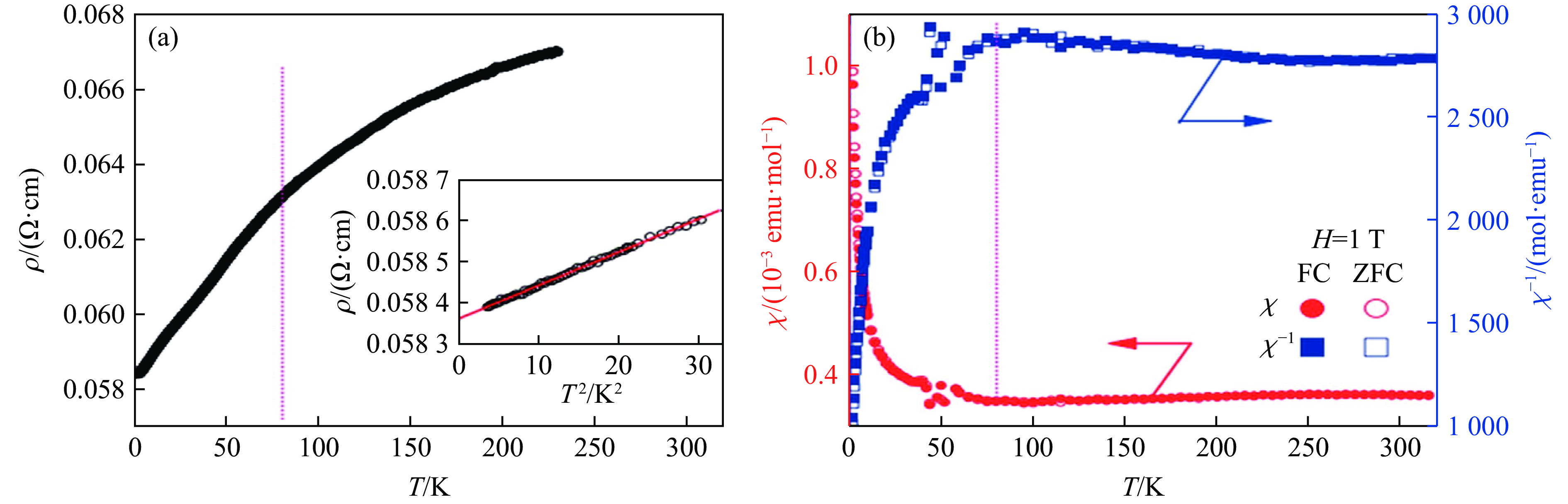
图14(a)显示了6H-BaFeO3的χ-T曲线[31]。6H-BaFeO3在低温下具有反铁磁性,其TN约为170 K。在高温下,6H-BaFeO3的χ-T曲线满足居里-外斯定律,然而,在170~230 K区间,χ−1-T曲线偏离了线性关系(见图14(a)中的插图)。由图14(a)中的插图可以得到,μeff约为2.39μB。如图14(b)所示,在升温的过程中,12M-BaFeO3在280 K处经历了从顺磁到反铁磁性的转变[33]。通过变温中子衍射数据计算各个位置上Fe离子的磁矩后发现,在500 K处,高温下部分Fe4+转变为Fe3+和Fe5+,即电荷发生了转移,对应于12R相(空间群为R
$ \overline{3} $ m)转变为12M相(空间群为C2/m)。在280 K处,Fe离子也发生了电荷转移,对应于反铁磁转变。在50 K处,发生了不同位置的Fe3+和Fe5+之间的转换,对应χ-T曲线在50 K下的转变。图14(c)显示了12M-BaFeO3在280~100 K区间和100 K下的磁结构[34],其中,Fe离子的磁矩基本上反平行排列。如图15(a)所示,3C-BaFeO3多晶体在低温下具有铁磁性[36],其TC为110 K,在此温度下,ZFC和FC曲线发生分离。利用式(2)拟合250~300 K区间的χ-T曲线,得到μeff为5.5μB,介于Fe3+的理论值5.916μB和Fe4+的理论值4.899μB之间。拟合得到的θ为163 K,说明近邻Fe离子的自旋是平行排列的。图15(b)显示了3C-BaFeO3单晶体的χ-T曲线,外加磁场分别平行于(110)面和(111)面。与多晶材料相比,单晶体的磁性更为复杂[37],在181 K处,3C-BaFeO3单晶体经历了自旋玻璃转变,在低温下经历了2个反铁磁性转变,TN分别为117和97 K。利用式(2)拟合210~300 K区间的χ-T曲线,得到μeff为6.15μB,更接近于Fe3+的理论值。拟合得到的θ为165.5 K,说明近邻Fe离子之间的铁磁相互作用占主导地位。Liu等[37]认为,正的外斯温度对应于3C-BaFeO3中旋转的铁磁平面内近邻Fe离子之间的相互作用,但整个晶体的自旋结构是反铁磁的。
-
在高氧压环境下合成的BaCoO3为2H相(图11(a))[38–42]。Ba(NO3)2和Co(NO3)2·6H2O在高温下反应,也可得到2H-BaCoO3多晶体[43]。利用高温高压方法(
1200 ℃、6.0 GPa)合成的BaCoO2.37基本上也是2H结构[44],但Ba离子的位置不同,如图16(a)所示,Ba占据2个不同位置,占据率分别为0.802和0.099。这一结构可称为2H'相,其c轴较短,导致近邻的Co-Co距离仅为2.07 Å,小于2H相中的Co-Co距离2.38 Å。将BaCO3和Co3O4在904 ℃下烧结可得12H结构的BaCoO2.6[45],该结构为六方结构,空间群为P63/mmc。图16(b)显示了它的晶体结构,其中,M4O15四聚体与MO4四面体以顶点上的O离子连接,但MO4四面体之间不连接。将BaCO3和Co3O4在1000 ℃下烧结可以得到5H结构的BaCoO2.74(图4(a))[46]。BaF2、BaCO3和Co3O4在氩气流和高温下可得BaCoO2.22单晶体[47],其晶体结构见图16(c)。BaCoO2.22单晶体为立方钙钛矿,但与标准结构略有区别,称为3C'相。Co离子不在CoO6八面体的中心位置,Co离子的占据率为1/8,Co―O距离不等,O―Co―O夹角不等于180°。将BaO2、CoO和Co2O3在高温下烧结,可得3C'-BaCoO2.22多晶体,并伴随少量CoO杂质[47]。图17(a)显示了2H-BaCoO3的多晶体和单晶体的ρ-T曲线[40],单晶体和多晶体均为半导体,但其电输运性质有明显差别。图17(a)中的插图显示了单晶体在160 K以上的电阻率ρ随着温度的变化,拟合得能隙Eg为0.061 eV。图17(b)显示了2H-BaCoO3单晶体在低温下的χ-T曲线(下标c和ab分别代表外磁场H平行c轴和ab平面),2H-BaCoO3单晶体在低温下具有二维反铁磁性,TN为15 K,与多晶体的结果[41–42]相似。通过拟合150 K以上的χ-T数据,得到μeff为1.76μB,与低自旋态Co4+的理论值1.73μB基本一致。θ为−25 K,表明近邻的Co离子之间的反铁磁相互作用占主导地位。
-
在高氧压环境下合成的BaNiO3为2H相(图11(a))[48]。在低自旋态的Ni4+中没有未成对的单电子[49],因此,2H-BaNiO3在低温下具有抗磁性。当M为Sc、Cu和Zn时,未形成BaMO3型化合物。
-
在常压、
1100 ℃的合成条件下,BaRuO3形成9R相[50–51];在高温高压合成条件下,BaRuO3形成4H、6H或3C相[52–55];在150 MPa、650 ℃的合成条件下,获得了BaRuO3的10H相单晶[56]。9R、4H、6H、3C和10H相的晶体结构见图11(b)、图8(a)、图3(a)、图1(a)和图11(e)。如图18(a)[54]所示,9R-BaRuO3在较高温度下为金属,在低温下变为绝缘体,其金属-绝缘体转变温度TMI约为118 K。4H-BaRuO3和6H-BaRuO3在实验温度范围内均为金属,在较高温区,ρ和T均不满足布洛赫线性关系,说明它们属于坏金属。如图18(a)中的插图所示,4H-BaRuO3在低温下的ρ-T关系满足式(3),其中n = 2。所以,4H-BaRuO3属于费米液体金属[54],ρ0 = 0.153 mΩ∙cm,剩余电阻比率(rRRR = ρ300 K/ρT→0)等于8.11。6H-BaRuO3在低温下的ρ-T关系满足式(3),其中n = 3/2,表明6H-BaRuO3在低温下具有非费米液体行为[54],ρ0 = 0.374 mΩ∙cm,rRRR = 7.24,所以6H相的ρ0比4H相的大,6H相的rRRR比4H相的小,说明6H相的金属性比4H相弱。6H相中,Ru与Ru之间通过八面体顶点O连接的比例更大,Ru2O9二聚体中Ru-Ru之间的距离更长,这可能是其金属性比4H相弱的原因。
图18(b)显示了BaRuO3的9R、4H和6H相的磁化率随温度的变化关系,由于ZFC和FC曲线一致,所以图中只显示ZFC结果。9R-BaRuO3在高温下具有短程反铁磁性,TN约为440 K,在低温下具有顺磁性[53]。4H-BaRuO3和6H-BaRuO3为电子交换增强的泡利顺磁体,这是由于晶胞中RuO6八面体共顶点连接比例增加,导致温度敏感性降低,但磁化率却大大提高[53–54]。它们的低温χ-T曲线满足
式中:A = (π2
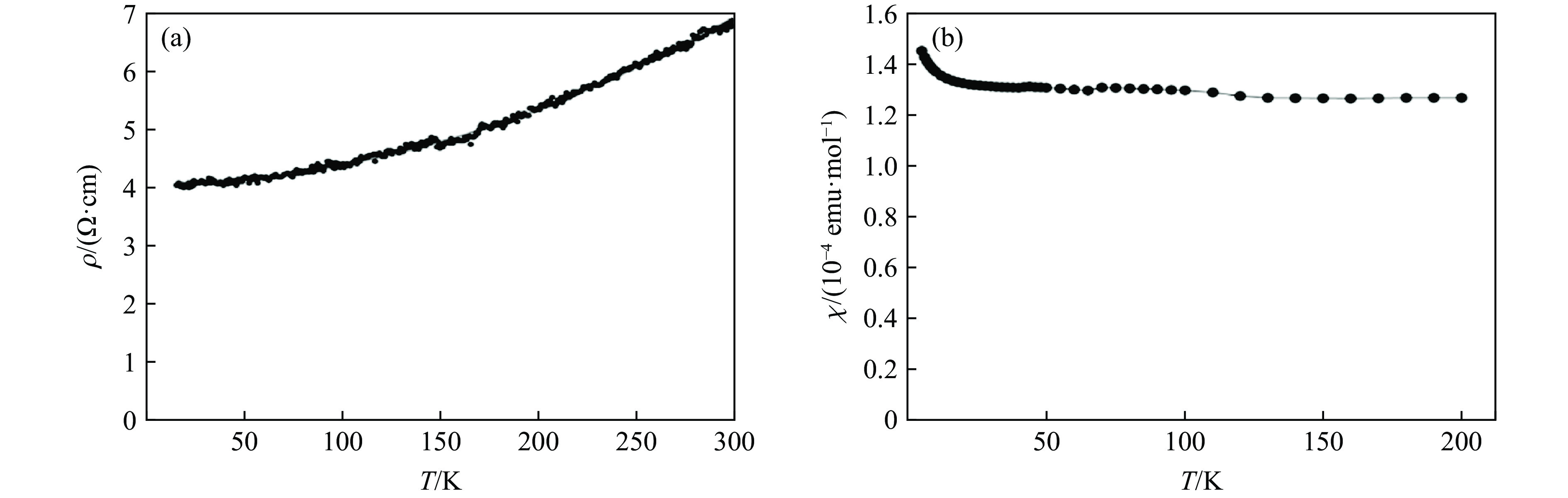
$k_{\mathrm{B}}^2 $ /6)[(N'(EF)/N(EF))2−(N"(EF)/N(EF))],N(EF)为费米面EF处每个原子的态密度,N'(EF)和N"(EF)分别为N(EF)对能量的一级和二级导数。4H-BaRuO3和6H-BaRuO3的θ分别为−6.7和−17.5 K,μeff分别为0.157μB和0.259μB,均比Ru4+的理论值2.83μB小得多,说明Ru离子缺失了部分局域磁矩,可能与Ru2O9二聚体中Ru-Ru的直接相互作用以及强自旋-轨道耦合作用有关。10H-BaRuO3属于顺磁性半导体[56],其χ-T曲线与4H-BaRuO3的相似,拟合实验数据得到的居里常数很小,说明10H-BaRuO3的顺磁有效磁矩很小,与4H-BaRuO3和6H-BaRuO3的结果相近。图19显示了3C-BaRuO3的ρ-T曲线和χ-T曲线。3C-BaRuO3属于弱的巡游铁磁性金属,TC约为60 K[55]。电阻率随温度变化曲线具有一个明显的转折点,对应居里温度。低温下,3C相的ρ与T的关系满足式(3),其中n≈2,说明它属于费米液体金属。3C相的ρ0 = 48.1 mΩ∙cm,rRRR = 2.82,与正交相的SrRuO3、CaRuO3相比,3C-BaRuO3的金属性较弱,这是由于3C-BaRuO3中Ru-O距离(2.003 Å)较长,导致近邻Ru离子之间的电子波函数交叠较少。利用居里-外斯定律对顺磁区间的χ-T曲线进行拟合,得到μeff = 2.509μB,与Ru4+的理论值2.83μB接近;θ = 64 K,略高于TC。尽管3C-BaRuO3中Ru-O-Ru夹角为180°,但较大的Ru-O距离使Ru4+的巡游性比正交相SrRuO3的巡游性更弱。
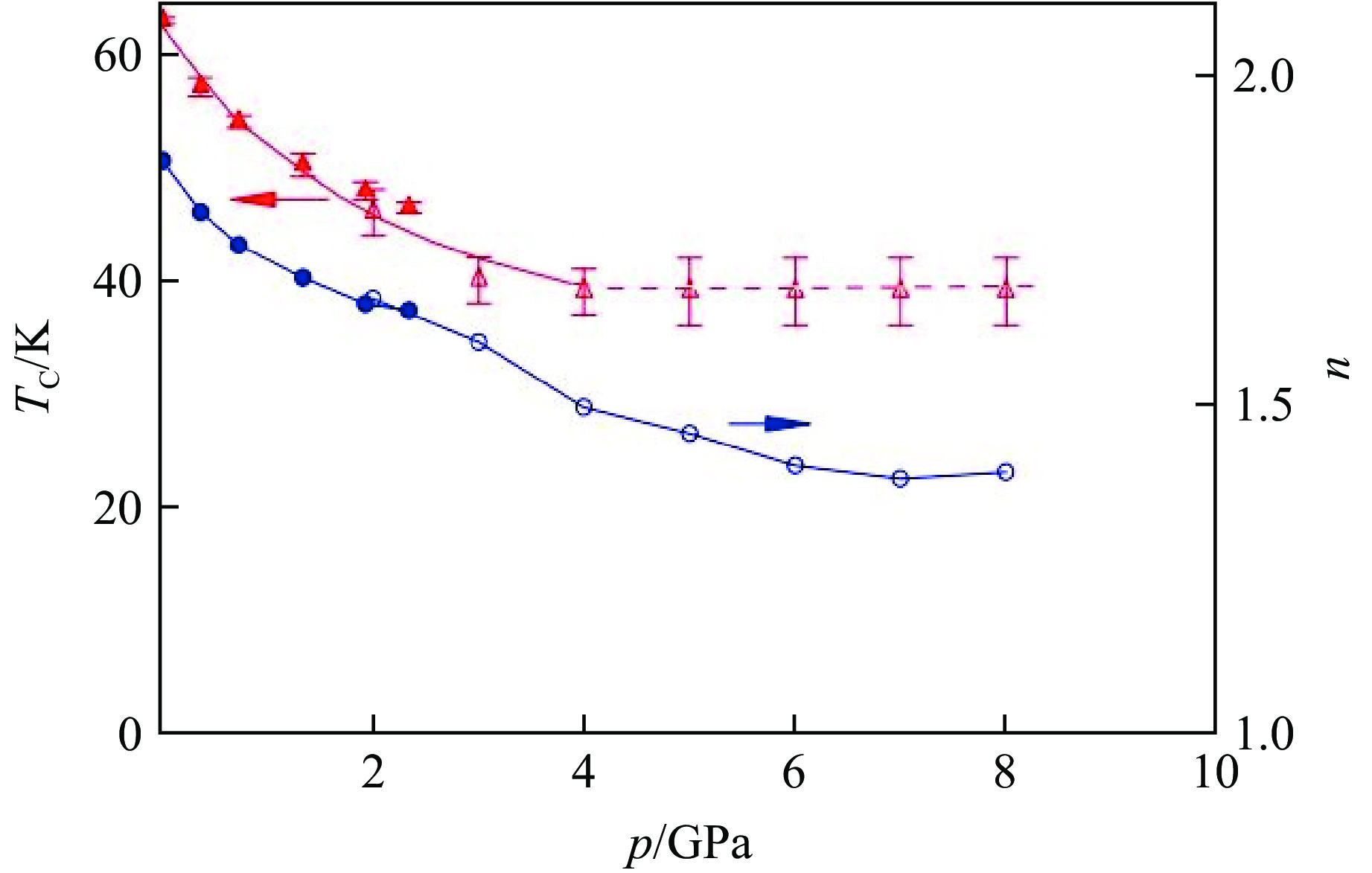
测量3C-BaRuO3在不同压力下的ρ-T曲线,通过计算dρ/dT得到TC随压力的变化[57]。如图20所示,3C-BaRuO3的TC随压力的增加而降低,从常压下的约60 K降低到4 GPa下的约40 K。当压力超过4 GPa,TC随压力增加基本不变,对应于铁磁性坍塌[57]。∂TC/∂p约为7.0 K/GPa[55, 57]。对于3C-SrRuO3(常压下的TC约为165 K),利用低温电性和磁性数据得到的∂TC/∂p分别为−6.2 和−5.7 K/GPa[58],∂ln TC/∂p = −3.5% GPa−1,与BaRuO3的结果接近,∂ln TC/∂p的值说明3C-SrRuO3和3C-BaRuO3属于弱的巡游铁磁体。对于3C-BaRuO3,利用式(3)拟合低温下的ρ-T曲线,指数n随压力的增加而减小,从常压下的1.85降低到铁磁性坍塌压力下的1.40,说明压力导致3C-BaRuO3从费米液体相转变为非费米液体相[57]。
-
在常压、
1100 ℃的合成条件下,BaIrO3形成畸变的单斜结构(空间群为C2/m)[59],一个晶胞包括12个BaIrO3化学式,沿c轴方向有6个BaO3层。由于离子的空间排列与9R结构类似,这个结构称为9M相。在高温高压合成条件下,BaIrO3形成另外2种畸变的单斜结构,称为5M和6M相,所属空间群分别为C2/m和C2/c,每个晶胞内分别包含10和12个BaIrO3化学式,合成压力分别为3.3和5.0 GPa[60–62]。6M相中离子的空间排列与6H相的类似,但5M相中离子的空间排列与5H相的并不类似。从离子的空间排列方面看,5M相介于4H与6H相之间。在25.0 GPa的合成压力下,BaIrO3形成四方钙钛矿(3C,空间群为I4/mcm)[63],离子的空间排列非常接近立方钙钛矿,IrO6八面体均共顶点连接。图21显示了BaIrO3各相的晶体结构。5M相中,2个M2O9二聚体共顶点连接,再与1个MO6八面体共顶点连接,二聚体与八面体沿c轴交替排列,BaO3平面的堆积序列为hchcc。如图22(a)[64]所示,9M-BaIrO3具有绝缘体性质,ρ-T曲线在TC附近有一个明显的转折点,在小于TC的区间,电阻率随温度的降低而迅速增加,说明其绝缘体特性随温度的降低更加显著。5M-BaIrO3和6M-BaIrO3在实验温度范围内均为金属,在较高的温度下ρ与T之间不是线性关系。5M-BaIrO3和6M-BaIrO3在低温下的ρ-T曲线满足式(3),分别拟合50和63 K之下的ρ-T曲线,得到它们的ρ0分别为3.218和1.588 mΩ∙cm,rRRR分别为2.147和2.519,说明6M相的导电性略好一些。5M-BaIrO3和6M-BaIrO3的n分别为2和5/3(图23(a)中插图),表明5M相是费米液体金属,而6M相在低温下具有非费米液体行为,可能处于量子临界点[63]。
如图22(b)所示,9M-BaIrO3表现出较弱的铁磁性,TC约为180 K,该磁现象是由Ir离子的自旋极化造成的[65],在小于TC的区间,ZFC和FC曲线的分离与快速零场冷却有关[66]。如图22(c)所示,9M-BaIrO3在5 K时具有较大的磁滞回线(图22(c)中回线向上进行了平移)。5M-BaIrO3具有弱铁磁性,TC约为50 K,它在5 K时具有磁滞回线,但磁化强度很小并且很难达到饱和。6M-BaIrO3具有顺磁性,磁化率在100~300 K范围内表现出微弱的温度依赖性,在较低温度下表现出轻微的增强,在5 K时无磁滞回线,磁化强度很小,但磁化强度与磁场之间并不是线性关系。9M-BaIrO3、5M-BaIrO3和6M-BaIrO3的μeff分别为0.186μB、0.346μB和0.364μB,均比Ir4+的理论值1.73μB小得多,这说明Ir离子缺失了部分局域磁矩,可能与Ir3O12三聚体或Ir2O9二聚体中Ir-Ir的直接相互作用以及强的自旋-轨道耦合作用有关。9M-BaIrO3、5M-BaIrO3和6M-BaIrO3的θ分别为162.9、−22.3和−64.9 K[64]。
图23显示了9M-BaIrO3在不同压力下的ρ-T曲线[67]和χ-T曲线[68],在实验温度范围内,其电阻率随压力的增加而增大,磁化率随压力的增加而减小。图23(a)和图23(b)中的插图分别显示了通过ρ-T曲线和χ-T曲线得到的TC随压力的变化关系。随着压力增加,9M-BaIrO3的TC逐渐减小,在实验压力范围内,TC与p基本为线性关系。利用低温电性和磁性数据得到的dTC/dp分别为−6.1和−17 K/GPa,两者存在较大的差别,可能与不同的实验条件、TC的定义等因素有关。
图24显示了3C-BaIrO3的ρ-T曲线和χ-T曲线[63]。3C-BaIrO3在低温时ρ-T关系满足式(3),ρ0 = 58.37 mΩ∙cm,rRRR约为1.147;n = 2,表明它是费米液体金属。3C-BaIrO3在较高温度下表现出与温度几乎无关的泡利顺磁性,χ-T曲线在85 K时出现最小值,这一温度对应于ρ-T曲线中斜率的变化。
-
在常压下合成的BaRh0.92O3为9M结构(图21(a))[69]。BaO2和RhO2混合物在6.0~6.5 GPa、
1175 ℃条件下形成了4H结构(图8(a))的BaRhO3[70]。在14.0~22.0 GPa的合成条件下,BaRhO3形成6M结构(图21(c))[71]。如图25(a)所示,4H-BaRhO3和6M-BaRhO3在低温下均具有交换增强的泡利顺磁性[71],其χ-T曲线符合式(4)。通过拟合实验数据,得到它们的θ分别为−13.5和−4.3 K,μeff分别为0.271μB和0.229μB,均比Rh4+的理论值1.73μB小很多,说明Rh离子缺失了部分局域磁矩。图25(b)显示了4H-BaRhO3和6M-BaRhO3在低温下的ρ-T曲线[71],4H相具有金属性,而6H相属于半导体。
-
在常压下合成的BaZrO3、BaNbO3、BaMoO3和BaHfO3均为立方钙钛矿[72–76],BaNbO3和BaMoO3为顺磁性金属[73–75],BaNbO3的μeff为0.12μB,θ为−2 K。在常压下合成的BaTcO3形成6H相(图3(a))[77]。在常压合成条件下,BaOsO3形成KSbO3型体心立方结构(bcc)[78];在约6 GPa的合成压力下,BaOsO3形成6H相[79–80](未给出具体的原子位置信息)。在17 GPa、
1600 ℃下,BaOsO3形成立方钙钛矿[80],为顺磁性材料,利用式(2)拟合χ-T曲线,得到μeff = 0.403μB,θ = 0 K。3C-BaOsO3在60 K经历了金属-半导体转变,利用式(3)拟合金属区域的ρ-T曲线,得到n = 2,ρ0 = 17.43 mΩ∙cm。在高氧压合成条件下,BaPtO3形成12层堆垛结构[81](未给出具体的原子位置信息)。在有催化剂的条件下,BaPtO3能形成立方钙钛矿[82]。当M为其他4d或5d过渡金属时,未形成BaMO3型化合物。 -
图26总结了BaMO3(M为过渡金属离子)所形成的多层堆积变体情况,其中红色为可以利用高温高压方法合成的材料,小括号显示了过渡金属离子六配位、+4价的离子半径[3]。在图26的灰色梯形区域,BaMO3易于形成六方钙钛矿。3d过渡金属离子半径较小,一般具有较多的价态,当M离子的平均价态小于+4时,造成O离子的含量少于3,可能形成BaMO3−δ。图26中的中括号表示所形成化合物的δ > 0.2。M以3d→4d→5d过渡金属离子顺序改变时,BaMO3形成六方钙钛矿时,M的数目逐渐减少,这是由于M离子的半径逐渐增大,容忍因子t发生变化。BaMO3能否形成六方钙钛矿或钙钛矿与M位离子的半径和价态密切相关。Ba2+(十二配位)和O2−(六配位)的离子半径分别为1.61和1.40 Å。依据式(1),当M离子的半径小于0.729 Å时,BaMO3的t 大于 1,可以形成六方钙钛矿。当M离子的半径大于0.579 Å时,M和O离子能够形成MO6八面体。第Ⅰ、Ⅱ和Ⅲ副族元素的离子半径较大、价态较小,所以未形成BaMO3型化合物。一些4d和5d过渡金属四价离子的半径介于0.579 与0.729 Å之间,但其BaMO3未形成六方钙钛矿结构,可能还与其他因素(合成条件、杂质含量等)有关。
-
在不同的合成压力范围内,BaMO3可以形成不同的多层堆积变体。图27总结了BaMO3(M = V, Cr, Mn, Ru, Os, Rh, Ir)多层堆积变体的合成压力范围,合成温度约在850~
1350 ℃区间,所标注的合成压力为近似值。BaVO3在0~15 GPa的合成压力范围内除5H相外没有形成其他结构,而BaCrO3在转变为钙钛矿之前可以形成4H相和6H相。当在O位掺入H时,BaVO3−xHx (0.3 ≤ x ≤ 0.8)在高温高压条件下(1 000 ℃,3和7 GPa)形成6H相,BaVO2.1H0.9在高温高压下形成了立方钙钛矿[83]。由图7可知,当合成温度超过1000 ℃时,BaCrO3在高温高压下不形成4H相。因此,BaVO3和BaCrO3主要的高压合成序列为5H→6H→3C。目前的实验结果表明,BaMnO3的高压合成序列为2H→9R→4H→6H,BaRuO3的高压合成序列为9R→4H→6H→3C,考虑两者在结构上的相似性,它们的高压合成序列可归并为2H→9R→4H→6H→3C。因此,当合成压力足够高时,BaMnO3也可以形成3C相。在较低的合成压力下,BaOsO3的结构比较复杂,BaOsO3在更高的合成压力下形成6H相和3C相。BaMO3(M = Rh, Ir)的高压相变序列与BaRuO3类似,其高压相中包含畸变的六方钙钛矿,BaRhO3在足够高的合成压力下也可能形成与BaIrO3类似的3C相。根据前述结果,图28总结了BaMO3中MO6八面体的连接情况随合成压力的演化,其中:箭头代表合成压力增加的方向,底部的数字代表MO6八面体共顶点连接所占的比例。在这类材料中,最典型的高压合成序列是2H→9R→4H→6H→3C。2H、9R、4H、6H和3C相为最简单的多层堆积变体:2H相中MO6八面体均为共面连接,9R相中M3O12三聚体共顶点连接,4H相中M2O9二聚体共顶点连接,6H相中M2O9二聚体和MO6八面体共顶点连接,3C相中MO6八面体均为共顶点连接。随着合成压力的增加,MO6八面体共顶点连接的比例逐渐增加。9R与9M相中离子的空间排列相近,MO6八面体共顶点连接的比例相同。因此,从离子的空间排列上,本文将9R相和9M相近似看成一种结构,同样将6H和6M相近似看成一种结构。MO6八面体共顶点连接的比例在4H、5H和5M相中分别为50%、60%和60%,介于9R/9M相的33.3%与6H/6M相的66.7%之间。一些钙钛矿材料,如在常压下合成的3C-BaTiO3、3C-BaFeO3等,相比于它们的六方钙钛矿,其MO6八面体均为共顶点连接,相当于共顶点连接的比例更大(100%)。由于合成压力增加和MO6八面体共顶点连接比例增大均可使BaMO3趋于形成类似的晶体结构,为了便于讨论问题,下文中对于同一M,将两者统称为合成压力增加。
-
图29(a)总结了BaMO3的单位晶胞体积V/Z,其中:Z为一个晶胞内包含的BaMO3化学式个数,9M、5M和6M相的Z分别为12、10和12;粗实线是按立方钙钛矿计算的理想值
$V{\text{/}}Z = {\left[ {2\left( {{r_{{{\text{M}}^{4+}}}}+{r_{{{\text{O}}^{2 - }}}}} \right)} \right]^3}$ ,${r_{{{\text{O}}^{2 - }}}} = 1.40$ Å,六方钙钛矿的V/Z基本上均比理想值大。除BaCrO3外,3C相的实验V/Z均比理想值小。3C-BaCrO3中O离子可能存在空位[10],Cr离子价态降低,晶胞体积比无空位时大,造成实验V/Z比理想值大[10]。对于同一结构,V/Z基本上随M离子半径的增加而增大。对于同一M,V/Z基本上随合成压力的增加而减小,说明合成压力的增加导致晶体结构中的空隙减小。在2个近邻的M-O多聚体之间,M离子与M离子之间通过O离子相连。对于9M、5M和6M相,M―O―M夹角在160°~170°之间;对于其他结构,M―O―M夹角基本上等于180°。图29(b)总结了BaMO3中近邻多聚体间M-M的距离dM-M,其中,粗实线是按立方钙钛矿计算获得的理想值
$ {d_{{{{\mathrm{M}} \text{-} {\mathrm{M}}}}}} = 2\left( {{r_{{{\text{M}}^{4+}}}}+{r_{{{\text{O}}^{2 - }}}}} \right) $ 。对于同一结构,dM-M基本上随M离子半径的增加而增大。对于同一M,dM-M基本上随合成压力的增加而增大。对于六方或三方晶系,轴比率c/a是一个重要的结构参数。为了便于比较,图29(c)总结了BaMO3六方钙钛矿的轴比率(c/a)/N,其中,N为晶胞内c轴方向BaO3层的个数。图中粗实线是基于2个正八面体共面连接计算的理想值
$( c{\text{/}}a)\text{/}N = 1{\text{/}}\sqrt 6 = 0.4082 $ 。如图29(c)中的插图所示,9M、5M和6M相为畸变的单斜结构(紫色边框),可以在其中划出一个类似六方结构的晶胞(绿色边框)。本文中将它们的晶胞参数用$ {a_{\text{H}}} = {b_{\text{H}}} = {{\sqrt {a_{\text{M}}^2+b_{\text{M}}^2} } \mathord{\left/ {\vphantom {{\sqrt {a_{\text{M}}^2+b_{\text{M}}^2} } 2}} \right. } 2} $ 和$ {c_{\text{H}}} = {c_{\text{M}}}\sin {\beta _{\text{M}}} $ (其中,aM、bM、cM和βM为单斜结构的晶胞参数)代替,9M、5M和6M相的N分别为6、5和6。6H相的(c/a)/N接近于理想值,其他相的(c/a)/N基本上均比理想值大。对于同一M,(c/a)/N基本上随着合成压力的增加而减小,说明合成压力的增加使BaMO3逐渐转变为钙钛矿。在六方钙钛矿的M-O多聚体中,M离子与M离子之间的距离较近。图29(d)总结了BaMO3六方钙钛矿多聚体内的M-M之间的距离
${d'_{{\text{M-M}}}} $ ,其中,粗实线是基于2个正八面体共面连接且M占据八面体中心计算的理想值${d'_{{\text{M-M}}}} = 2\left( {{r_{{{\text{M}}^{4+}}}}+{r_{{{\text{O}}^{2 - }}}}} \right){\text{/}}\sqrt 3 $ 。对于BaMO3六方钙钛矿,多聚体内的${d'_{{\text{M-M}}}} $ 均大于理想值。一方面,它们的(c/a)/N基本上比理想值大,说明c轴相对更长;另一方面,在多聚体内,M离子与M离子之间存在库仑排斥作用,导致${d'_{{\text{M-M}}}} $ 更大。对于同一M,${d'_{{\text{M-M}}}} $ 基本上随着合成压力的增加而增大,说明M离子和M离子之间的库仑排斥作用增强。图29(e)和图29(f)显示了BaMO3中Ba-O之间的平均距离
${\bar d_{{\text{Ba-O}}}}$ 和M-O之间的平均距离${\bar d_{{\text{M-O}}}}$ ,粗实线分别为理想值${\bar d_{{\text{Ba-O}}}} = {r_{{\text{B}}{{\text{a}}^{2+}}}}+{r_{{{\text{O}}^{2 - }}}}$ 和${\bar d_{{\text{M-O}}}} = {r_{{{\text{M}}^{4+}}}}+{r_{{{\text{O}}^{2 - }}}}$ 。对于同一结构,实验得到的${\bar d_{{\text{Ba-O}}}}$ 和${\bar d_{{\text{M-O}}}}$ 基本上随M离子半径的增加而增大。对于同一M,${\bar d_{{\text{Ba-O}}}}$ 基本上随合成压力的增加而减小,而${\bar d_{{\text{M-O}}}}$ 基本上随合成压力的增加而增大。 -
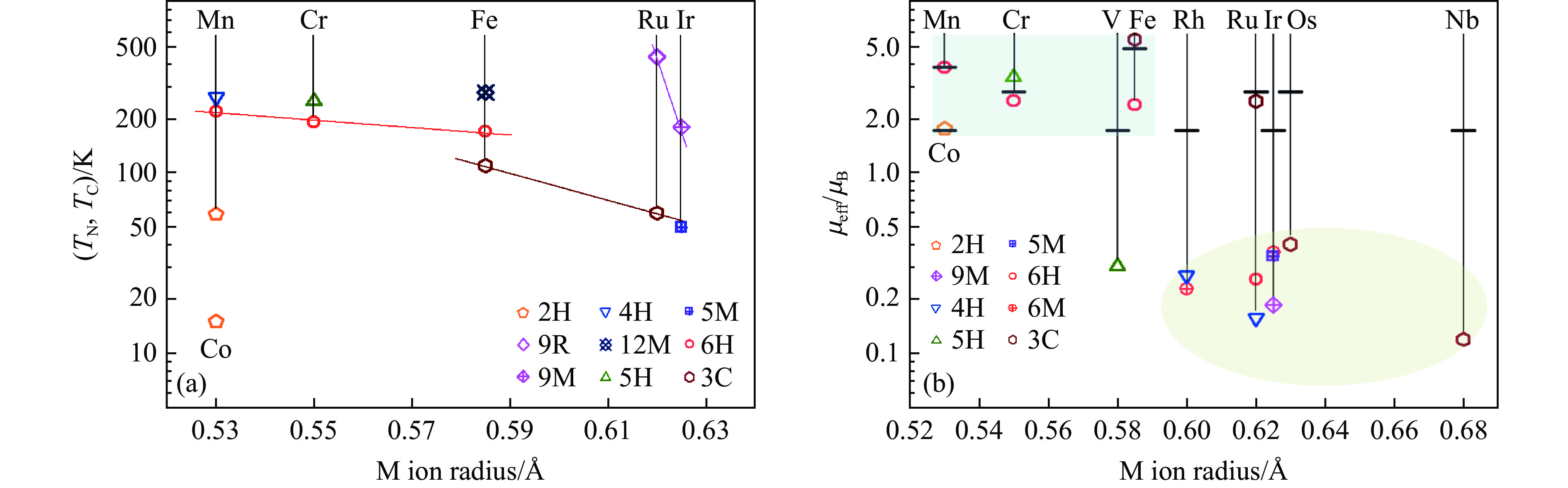
从图26中可以看出,在常压下利用固态反应方法合成的BaMO3多层堆积变体的结构与M离子(六配位、+4价)的半径密切相关:对于3d过渡金属,BaMO3多层堆积变体的相序列为2H相(M = Ni, Co, Mn; rM 为 0.48~0.53 Å)→5H相(M = V, Cr; rM 为 0.48~0.53 Å)→6H相(M = Fe, Ti; rM 为0.585~0.605 Å);对于4d和5d过渡金属,随着M离子半径的增加,BaMO3多层堆积变体的相序列为9R/9M相(M = Rh, Ru, Ir; rM 为0.6~0.625 Å)→6H相(M = Tc;rM 为 0.645 Å)→3C相(M = Mo, Nb, Hf, Zr; rM为 0.65~0.72 Å)。因此,当M离子半径增大时,常压下合成的BaMO3趋于高压高温相。
依据式(1),B位离子半径增大导致容忍因子t减小。如前所述,合成压力增大导致六方钙钛矿转变为钙钛矿,六方钙钛矿的t > 1,钙钛矿的t ≈ 1,即合成压力增大导致t减小。所以,从t的变化趋势上看,M离子半径增加与合成压力增大的作用相似。这与图27的结果相似,M离子半径增加与合成压力增大使BaMO3趋于形成类似的结构。当B为主族元素时,也有类似的情况,例如:BaSiO3在较高的合成压力下可以形成9R、6H和3C相,合成压力分别为27.9、48.5和141 GPa[84–85];BaGeO3在较低的合成压力下便可以形成9R、4H和6H相,合成压力分别为9.5、12和17.4 GPa[86];BaSnO3在常压条件下形成3C相[87]。当合成压力相同,如20 GPa,随着B离子半径增大(B为六配位时,Si4+、Ge4+和Sn4+的半径分别为0.40、0.53和0.69 Å),BaSiO3、BaGeO3和BaSnO3分别形成9R、6H和3C相。对于ABX3多层堆积变体,A离子半径减小导致t减小,与合成压力增大的作用相似,例如:对于Ba1−xSrxRuO3,当Sr含量增多(Sr为十二配位时,Sr2+半径为1.44 Å)或合成压力增大,都从六方钙钛矿转变为钙钛矿[88]。所以,A离子半径减小与B离子半径增大所起到的作用类似,均使ABX3转变为合成压力增加时的晶体结构。
对于同一M,在不同的合成条件下,BaMO3可形成不同的晶体结构,如果只按离子半径计算容忍因子t则无法反映出这些相之间的差异。因此,采用实验的晶体结构数据得到的
${\bar d_{{\text{Ba-O}}}}$ 和${\bar d_{{\text{M-O}}}}$ (图29(e)和图29(f))计算了BaMO3各相的“容忍因子”,即图30显示了t'随M离子半径的变化关系,其中,粗实线为根据式(1)和离子半径计算获得的t。对于BaMO3六方钙钛矿,实验得到的t'基本上都小于t,并且都大于1。对于同一结构,随着M离子半径的增大,t'基本上逐渐减小。对于同一M,随着合成压力增加,t'基本上逐渐减小。因此,对于t',M离子半径增大与合成压力增加起到了类似的作用。
-
当M为3d过渡金属离子时,除BaVO3外,BaMO3多为半导体或绝缘体,这是由于3d电子的局域性较强,导致BaMO3的导电性较差。当M为4d或5d过渡金属离子时,电子的巡游性较强,BaMO3多为金属;当材料中存在M3O12三聚体时,BaMO3的导电性较差,如9R-BaRuO3、10H-BaRuO3、9M-BaIrO3等。图31总结了BaMO3(M = V, Rh, Ru, Ir, Os)的剩余电阻率ρ0和剩余电阻比率rRRR。对于同一M,随着合成压力增加,ρ0基本上逐渐增大,而rRRR基本上逐渐减小,说明含有M2O9二聚体的六方钙钛矿的低温电阻率比钙钛矿的小,即前者的导电性比后者的好。而且,M2O9二聚体中M-M之间的距离(图29(d))比相应金属单质中的更小。因此,对于4d或5d过渡金属离子,M2O9二聚体中M离子与M离子间的直接相互作用更有利于电输运。
-
对于BaMO3钙钛矿,M离子之间通过八面体顶点的O离子产生交换作用,M―O―M夹角基本上等于180°。除3C-BaRuO3和3C-BaFeO3外,BaMO3钙钛矿均具有弱磁性。在六方钙钛矿中,M离子之间有2种连接方式:(1) 近邻多聚体中M离子通过八面体顶点的O离子相连,M―O―M夹角基本等于180°(六方和三方结构)或160°~170°(单斜结构);(2) 多聚体内M离子之间距离较近,M―O―M夹角小于90°。多聚体内M离子的自旋为平行或反平行排列,而多聚体间的M离子自旋一般反平行排列,所以材料在低温下多为反铁磁性或弱铁磁性。图32(a)总结了BaMO3的磁有序温度,包括奈尔温度TN和居里温度TC。对于同一结构,磁有序温度基本上随着M离子半径的增加而减小。对于同一M,除了2H相外,磁有序温度基本上随着合成压力的增加而减小。因此,对于改变磁有序温度,M离子半径增大与合成压力增加起到了类似的作用。由前面的结果可知,这类材料的磁有序温度随压力的增加而减小,如3C-BaRuO3、3C-SrRuO3、9M-BaIrO3等[57–58, 67–68]。
图32(b)总结了BaMO3的顺磁有效磁矩μeff,其中粗实线是理论值
$ {\mu _{{\text{eff}}}} = 2\sqrt {S\left( {S+1} \right)} $ (S为总自旋量子数)。Fe4+离子处于高自旋态。当M为3d过渡金属离子,实验得到的μeff与理论值基本接近(图32(b)中左上部阴影区),仅5H-BaVO3的μeff比理论值小得多。当M为4d和5d过渡金属离子,实验得到的μeff比理论值小得多(图32(b)中右下部阴影区),仅3C-BaRuO3的μeff与理论值基本接近,原因可能是M离子内的自旋-轨道耦合和多聚体中M离子之间的反铁磁交换作用导致电子巡游性增强。类似的现象存在于3C-SrIrO3和Ruddlesden-Popper结构的Sr2IrO4、Sr3Ir2O7等材料中,其μeff分别为1.17μB、0.05μB和0.69μB[88–90]。电子的巡游性增强可以提高材料的电输运性质,所以这些μeff比较小的材料的导电性(见图31)较好。综合前述BaMO3晶体结构参数的演化情况,对于同一M,当合成压力增加时,单位晶胞体积V/Z逐渐减小,说明结构中的空隙逐渐减少;Ba-O之间的平均距离
${\bar d_{{\text{Ba-O}}}}$ 逐渐减小,M-O之间的平均距离${\bar d_{{\text{M-O}}}}$ 逐渐增大,说明原子的空间位置分布发生了改变。如果BaMO3中MO6八面体都是正八面体且M离子位于八面体中心,则同一M的多层堆积变体应具有相同的V/Z、${\bar d_{{\text{Ba-O}}}}$ 和${\bar d_{{\text{M-O}}}}$ ,但六方钙钛矿为畸变的MO6八面体,M-O多聚体中M离子之间的库仑排斥作用使M离子偏离八面体中心,导致M离子之间出现较大空隙。高压导致六方钙钛矿中的畸变程度降低、空隙减小,六方钙钛矿逐渐转变为立方钙钛矿。对于同一结构,“容忍因子”t'和磁有序温度(TN、TC)随M离子半径的增加而减小,可以看出,M离子半径的增加与合成压力的增大产生了相似的效果。 -
本文对钙钛矿氧化物BaMO3(M为过渡金属离子)的高压合成、晶体结构和物理性质方面的研究工作进行了系统的总结,对相应的实验结果展开了详细讨论。对于钙钛矿和简单结构的六方钙钛矿,通过已有的实验数据计算了它们的晶胞参数和原子间距,获得了晶体结构和物理性质随M离子半径或合成压力的演化规律。基于实验获得的平均原子间距计算了“容忍因子”t',给出了t'随合成压力增加的大致变化情况,探讨了六方钙钛矿变为钙钛矿的可能过程。最后,总结了BaMO3的物性参数(包括剩余电阻率、剩余电阻比率、磁有序温度、顺磁有效磁矩等),得到了它们随M离子半径或合成压力的演化情况。
目前,尚未通过实验得到BaMO3(M = Pd, Ta, W, Re)的钙钛矿结构,可以通过改变实验条件(温度、压力等)获取这些材料并研究其物理性质。BaMnO3、BaTcO3和BaRhO3具有六方钙钛矿结构,但目前尚未获得它们的钙钛矿相,根据实验规律,可以推测出它们大致的合成压力、晶体结构参数和磁有序温度。此外,人们研究了6H-BaOsO3和12H-BaPtO3,但还未获得其具体的结构信息和物理性质,需要开展进一步的实验工作。M为4d和5d过渡金属离子时,BaMO3的顺磁有效磁矩比理论值小得多,对该现象尚未给出合理的物理机制。未来,对上述内容的研究可以完善这一体系的实验结果,支撑和发展本文所获得的结构和物性演化规律,为继续拓展钙钛矿氧化物的相关研究提供有效的实验数据。
钙钛矿氧化物BaMO3(M为过渡金属)的晶体结构和物理性质
Crystal Structure and Physica Properties of Perovskite Oxide BaMO3 (M Being Transition Metal)
-
摘要: 钙钛矿氧化物BaMO3(M为过渡族金属)具有复杂的晶体结构和物理性质,本文系统地总结了BaMO3的研究进展,重点关注在 M 元素变化过程中晶体结构和物理性质的演化,以及高压调控下的结构相变、电输运性质和磁学性质的变化,讨论了M离子半径及合成压力对六方钙钛矿到钙钛矿演化过程的影响,同时对该领域中一些问题做了展望,探讨了这一体系可能出现的新的原子组合和结构,相应材料可能具有的新特性和科学意义。Abstract: The perovskite oxide BaMO3 (M being transition metal) has a complex crystal structure and physical properties. This article systematically summarizes the research progress, focusing on the evolution of crystal structure and physical properties during the M element change process, as well as the structural phase transition, electrical transport properties, and magnetic properties regulation under high-pressure. The influence of M ion radius and synthesis pressure on the evolution process from hexagonal perovskite to perovskite is discussed, and some issues in this field are also discussed. The possible new atomic combinations and structures in this system, as well as the new characteristics and scientific significance of these corresponding materials, are discussed.
-

-
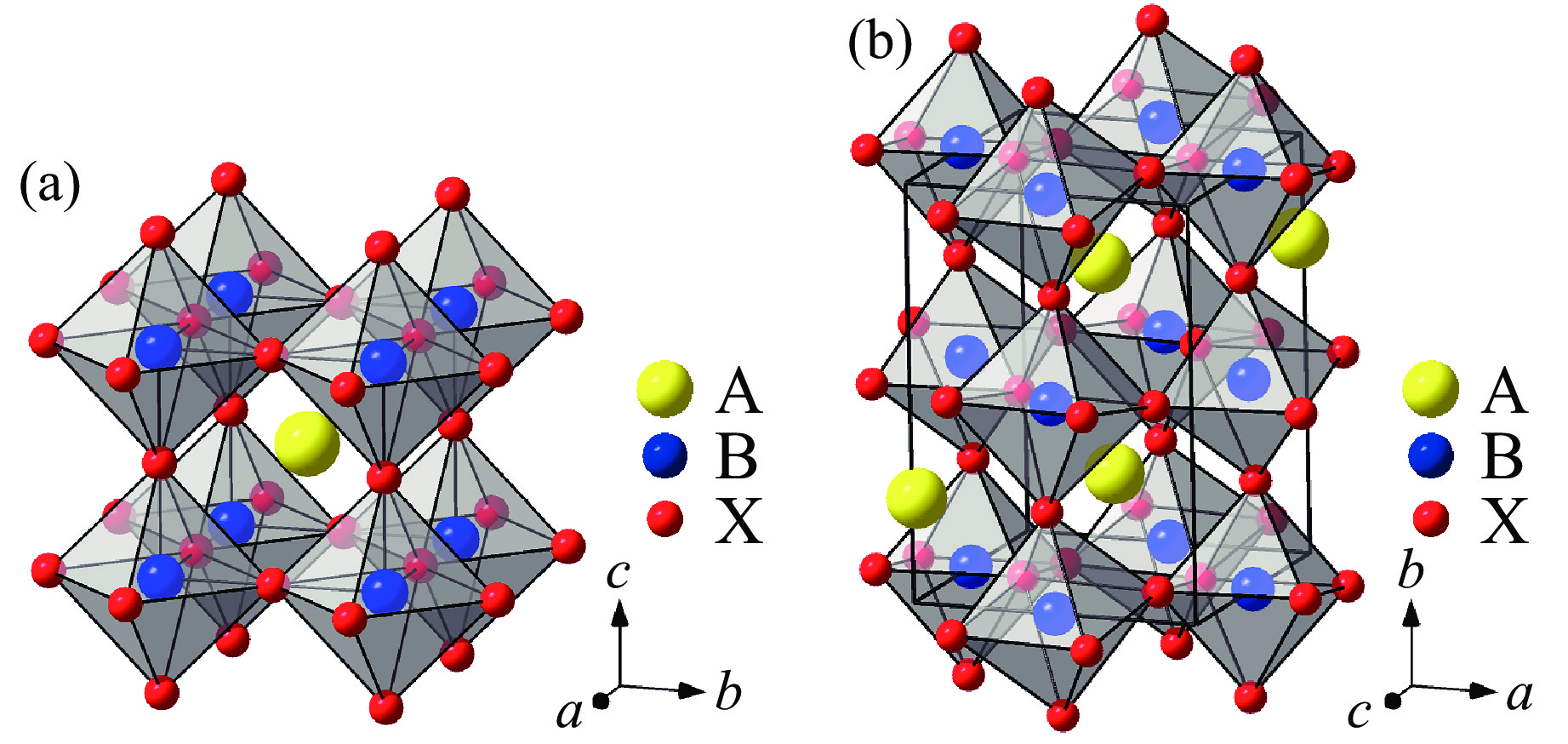
图 1 (a) 立方和(b) 正交钙钛矿的晶体结构示意图
Figure 1. Schematic views of the crystallographic forms of (a) cubic and (b) orthorhombic perovskite
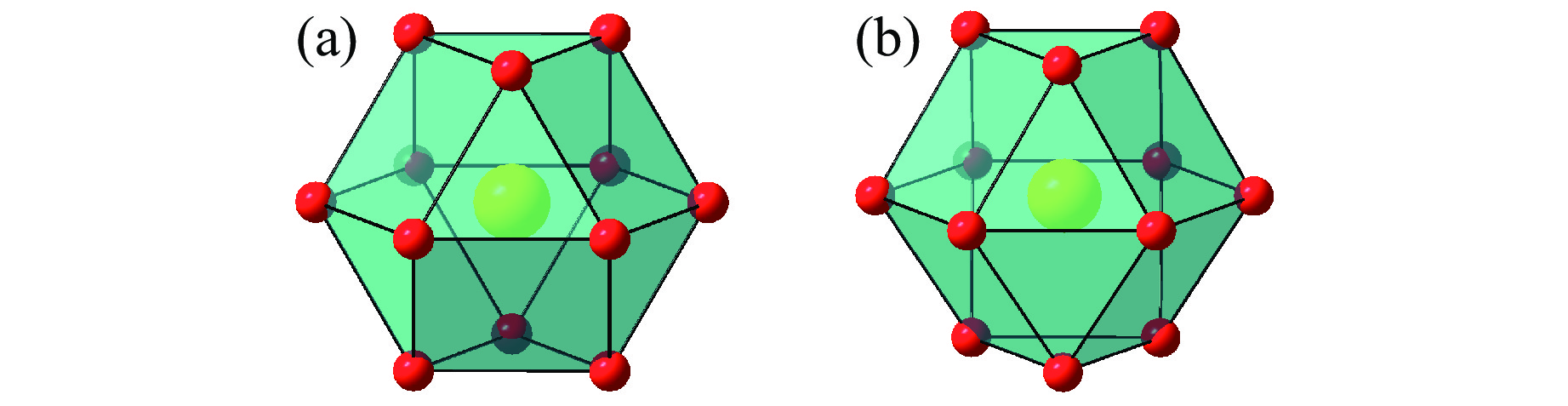
图 2 BX6八面体(a)共顶点连接和(b)共面连接时A-X配位示意图
Figure 2. Schematic views of A-X coordinations while BX6 octahedrons are connected by (a) vertex and (b) plane
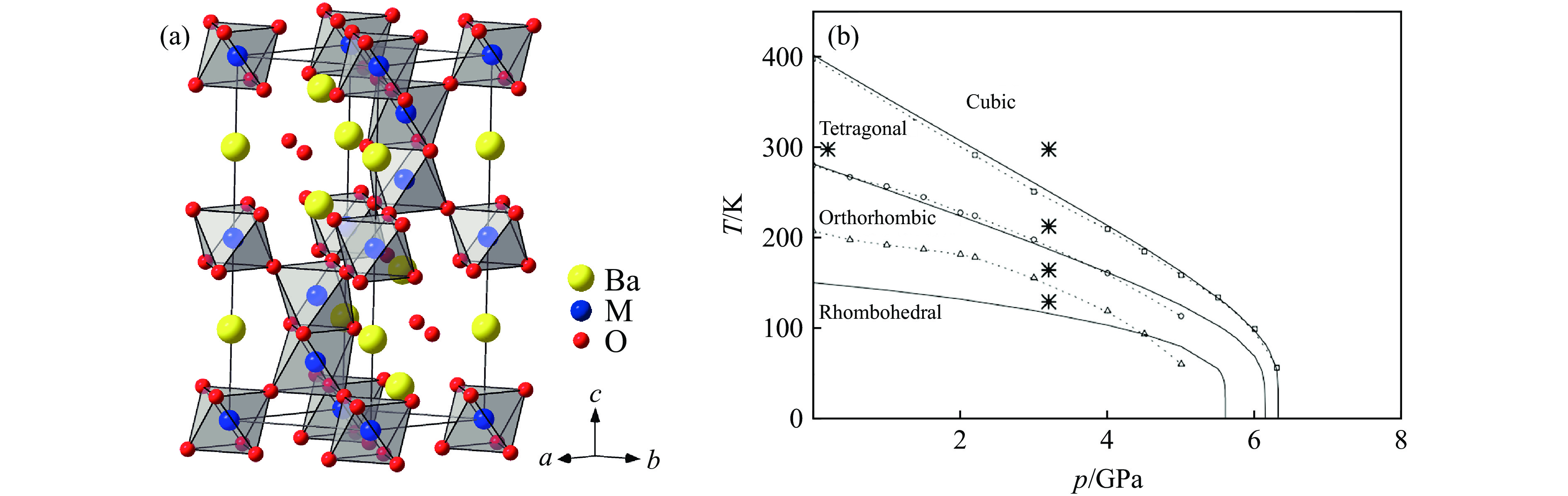
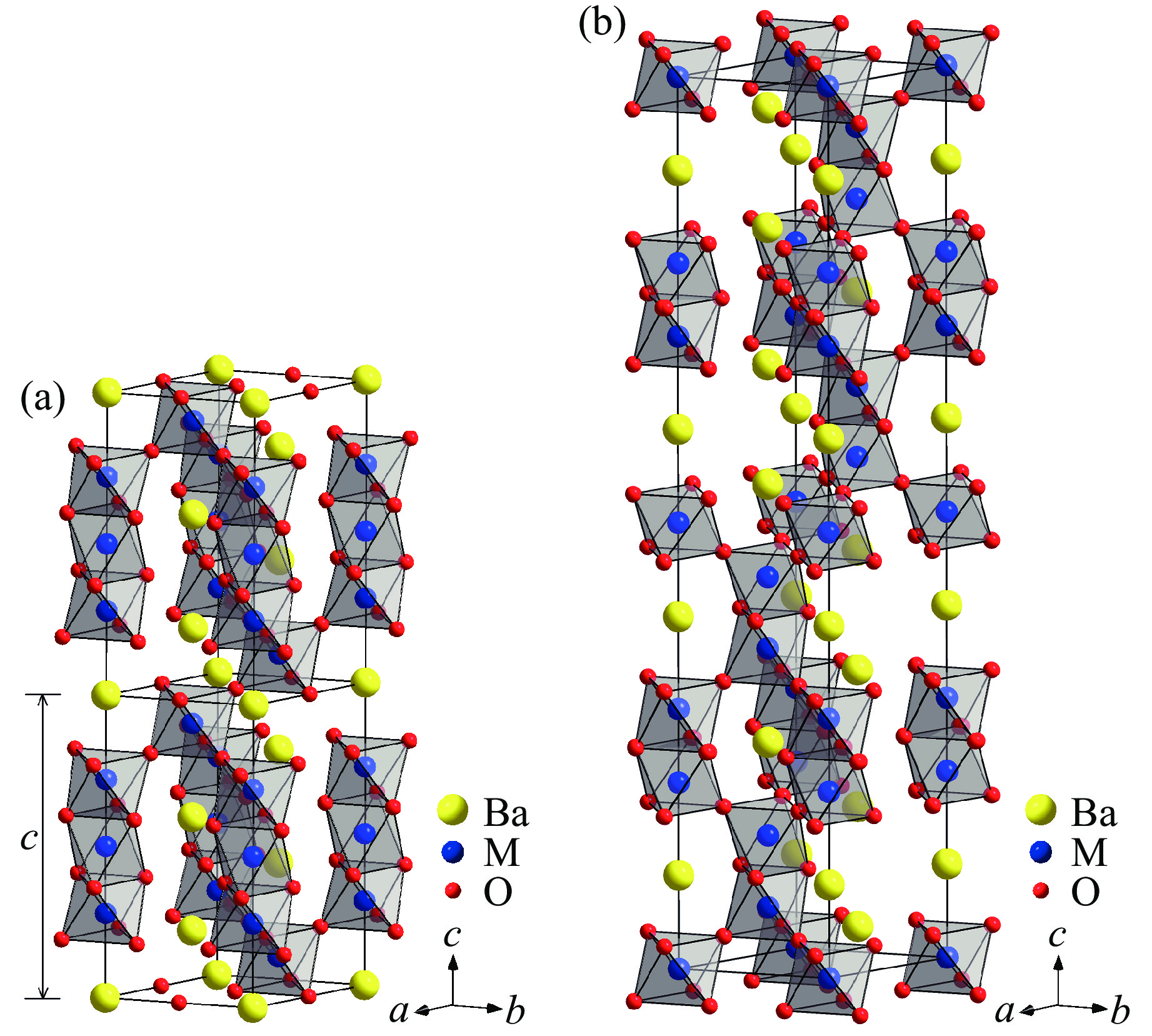
图 4 (a) 5H-BaMO3和(b) 14H-BaMO3的晶体结构示意图
Figure 4. Schematic views of crystal structure of (a) 5H-BaMO3 and (b) 14H-BaMO3
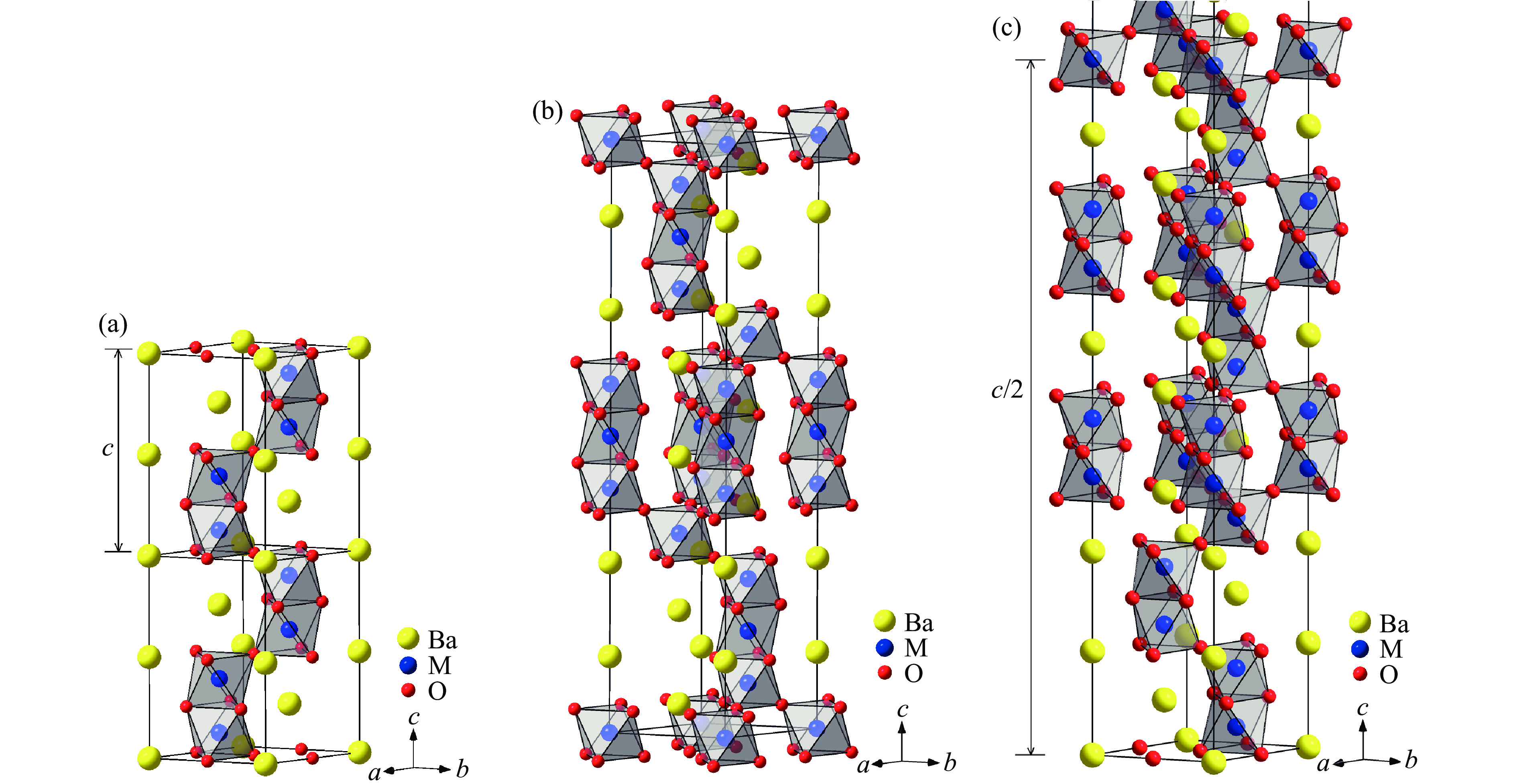
图 8 (a) 4H-BaMO3、(b) 12R-BaMO3和(c) 27R-BaMO3的晶体结构示意图
Figure 8. Schematic diagram of crystal structure of (a) 4H-BaMO3, (b) 12R-BaMO3, and (c) 27R-BaMO3
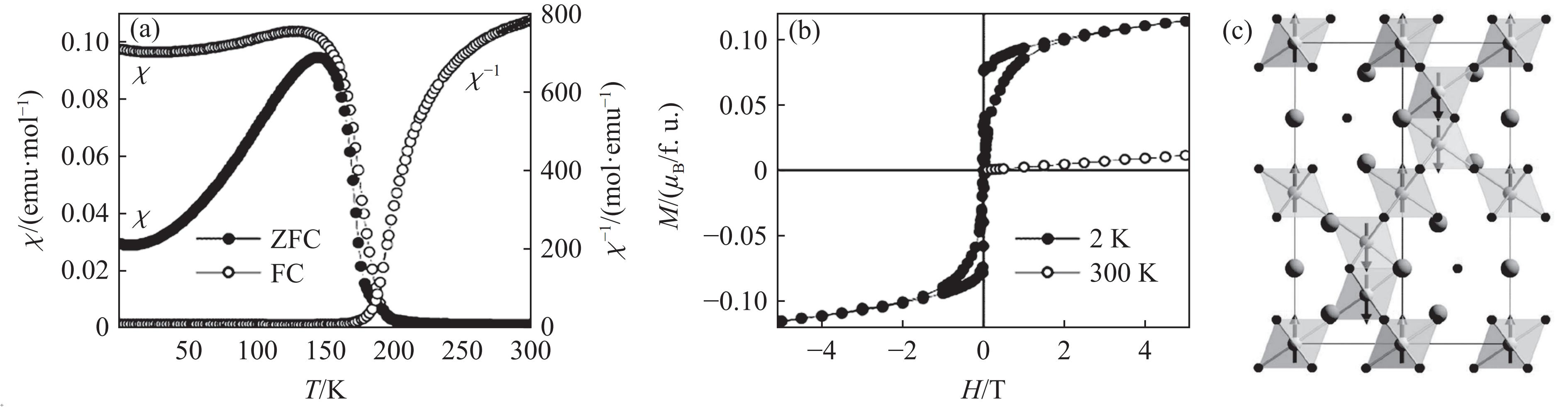
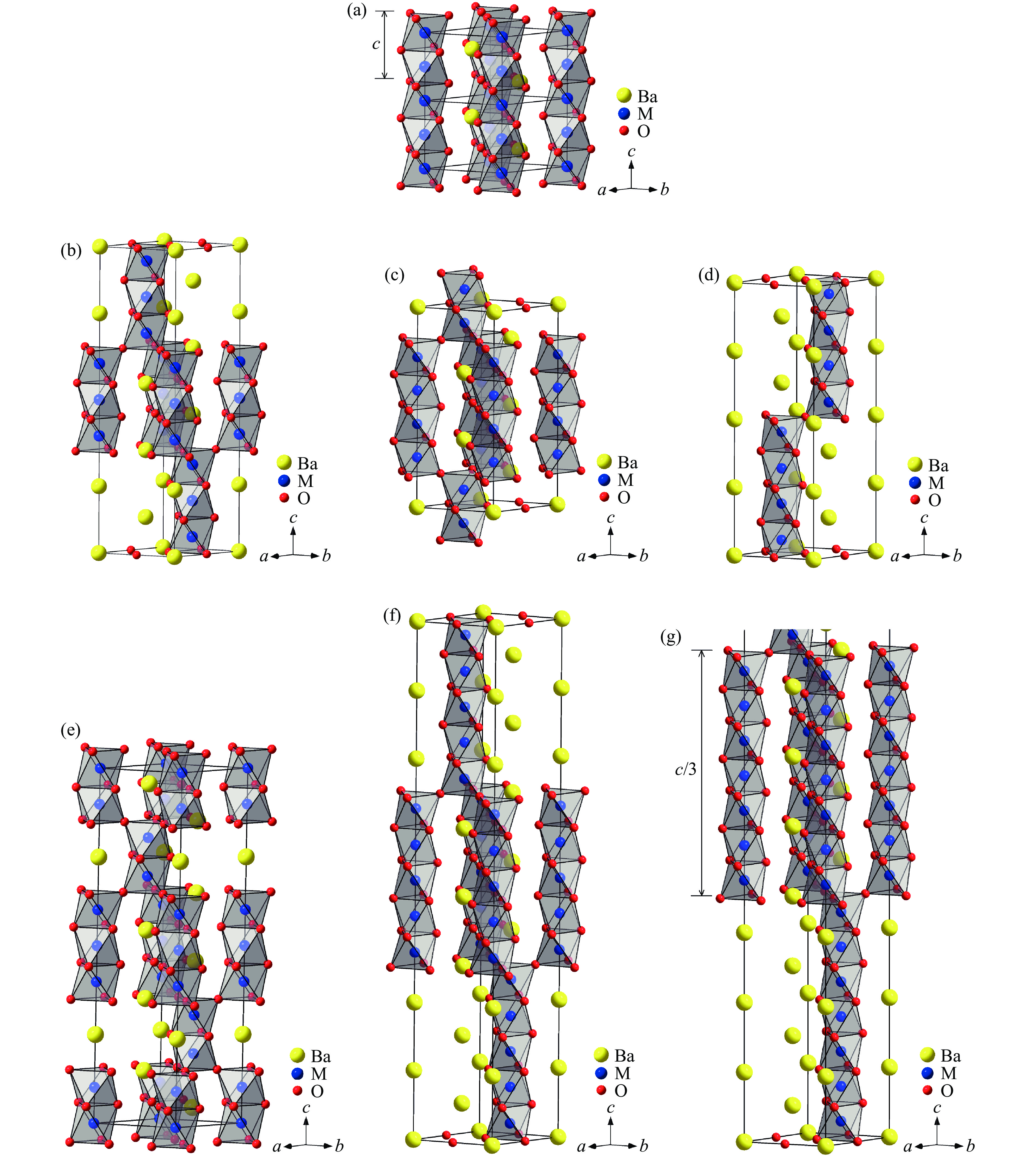
图 11 (a) 2H-BaMO3、(b) 9R-BaMO3、(c) 6H'-BaMO3、(d) 8H-BaMO3、(e) 10H-BaMO3、(f) 15R-BaMO3和(g) 21R-BaMO3的晶体结构示意图
Figure 11. Schematic diagrams of crystal structures of (a) 2H-BaMO3, (b) 9R-BaMO3, (c) 6H'-BaMO3, (d) 8H-BaMO3, (e) 10H-BaMO3, (f) 15R-BaMO3, and (g) 21R-BaMO3
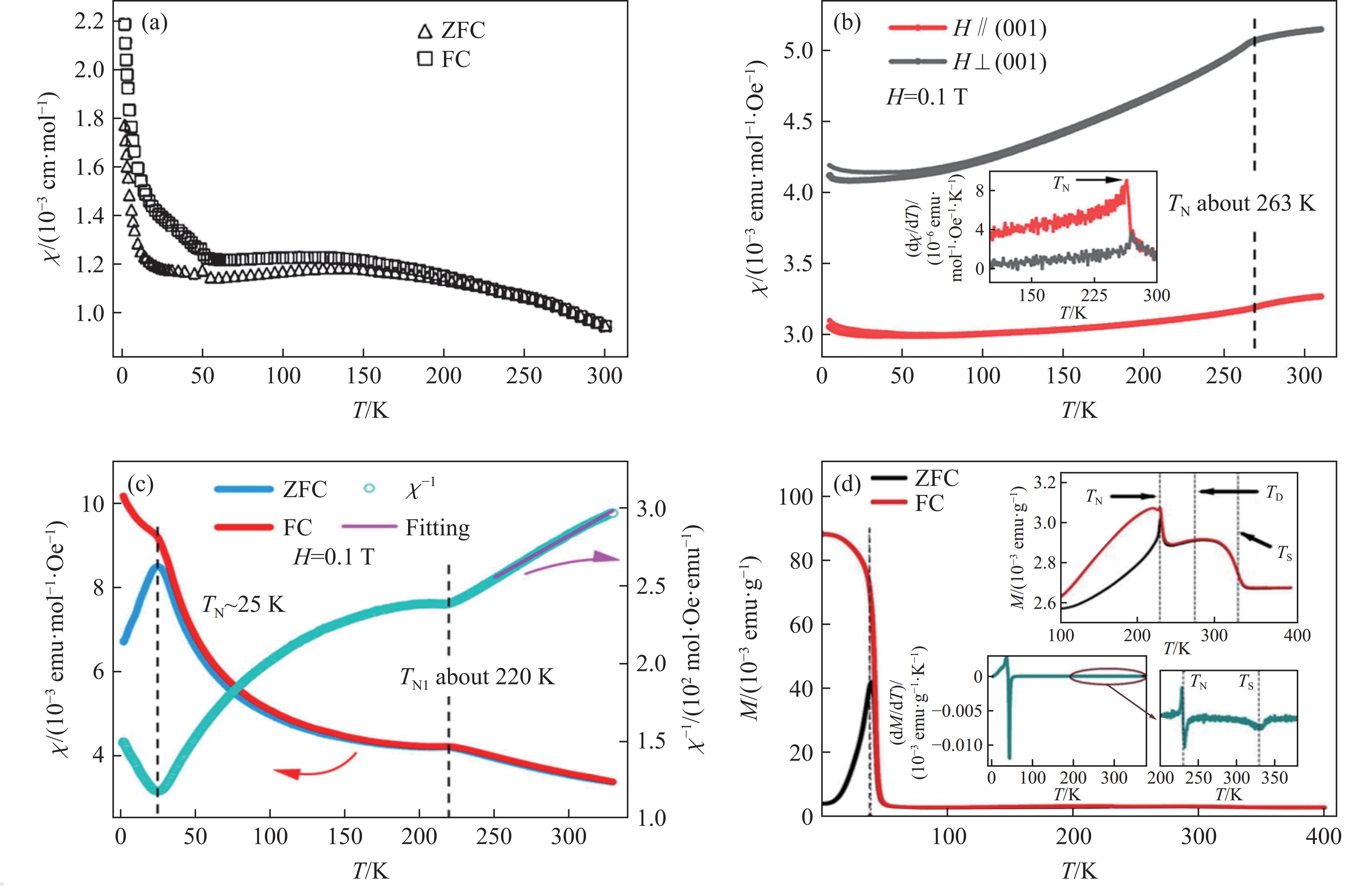
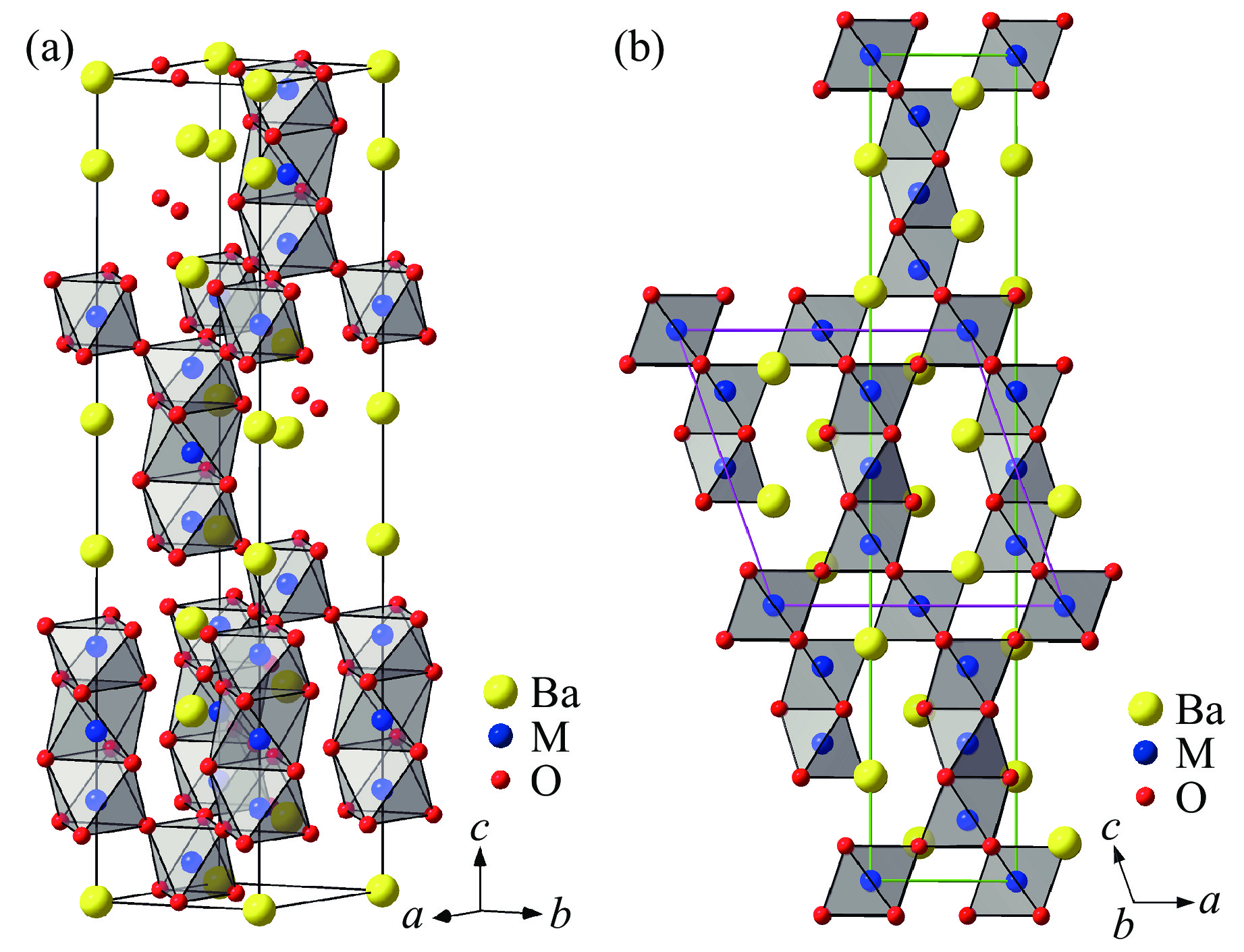
图 13 (a) “12R”-BaMO3和(b) 12M-BaMO3的晶体结构示意图
Figure 13. Schematic diagram of crystal structure of(a) “12R”-BaMO3 and (b) 12M-BaMO3
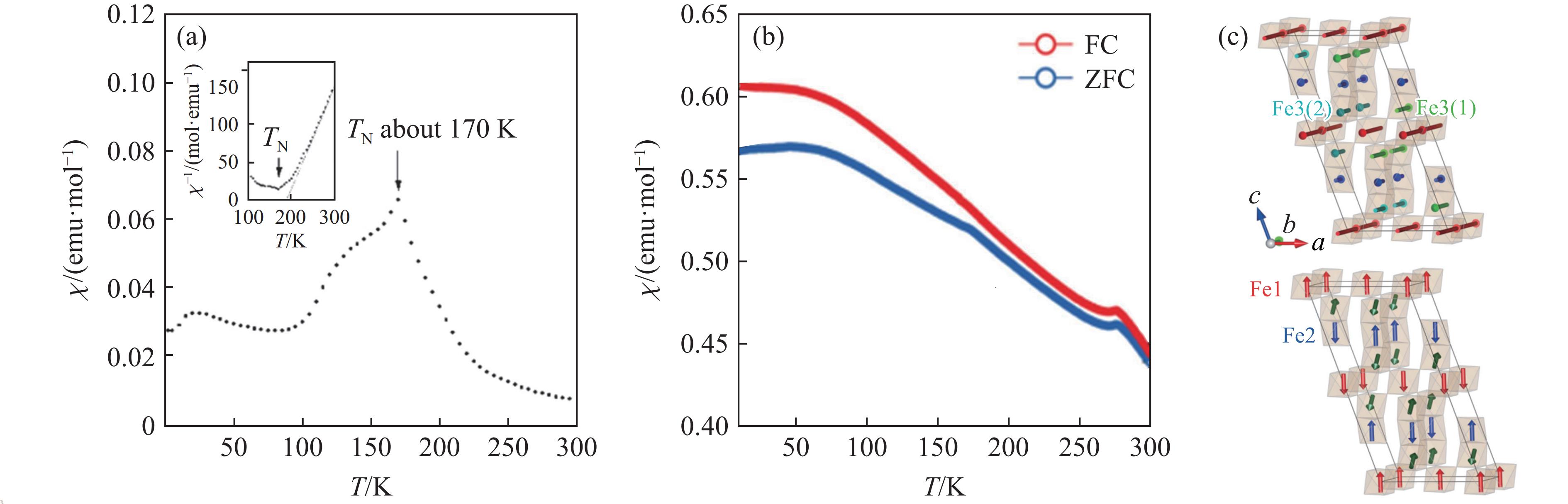
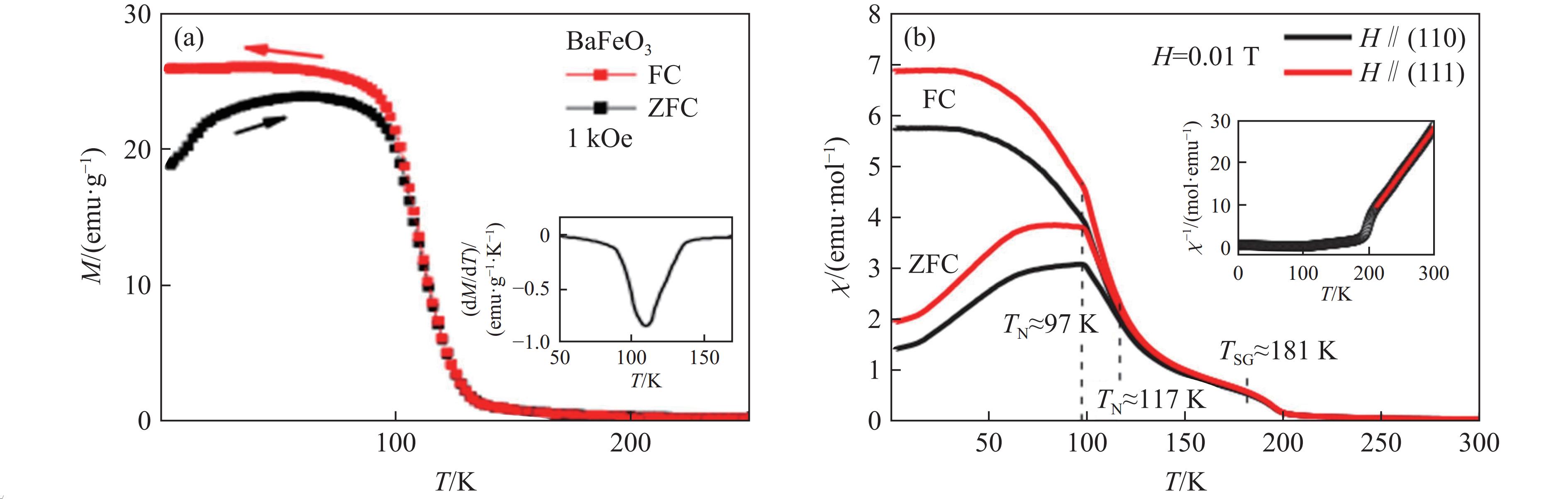
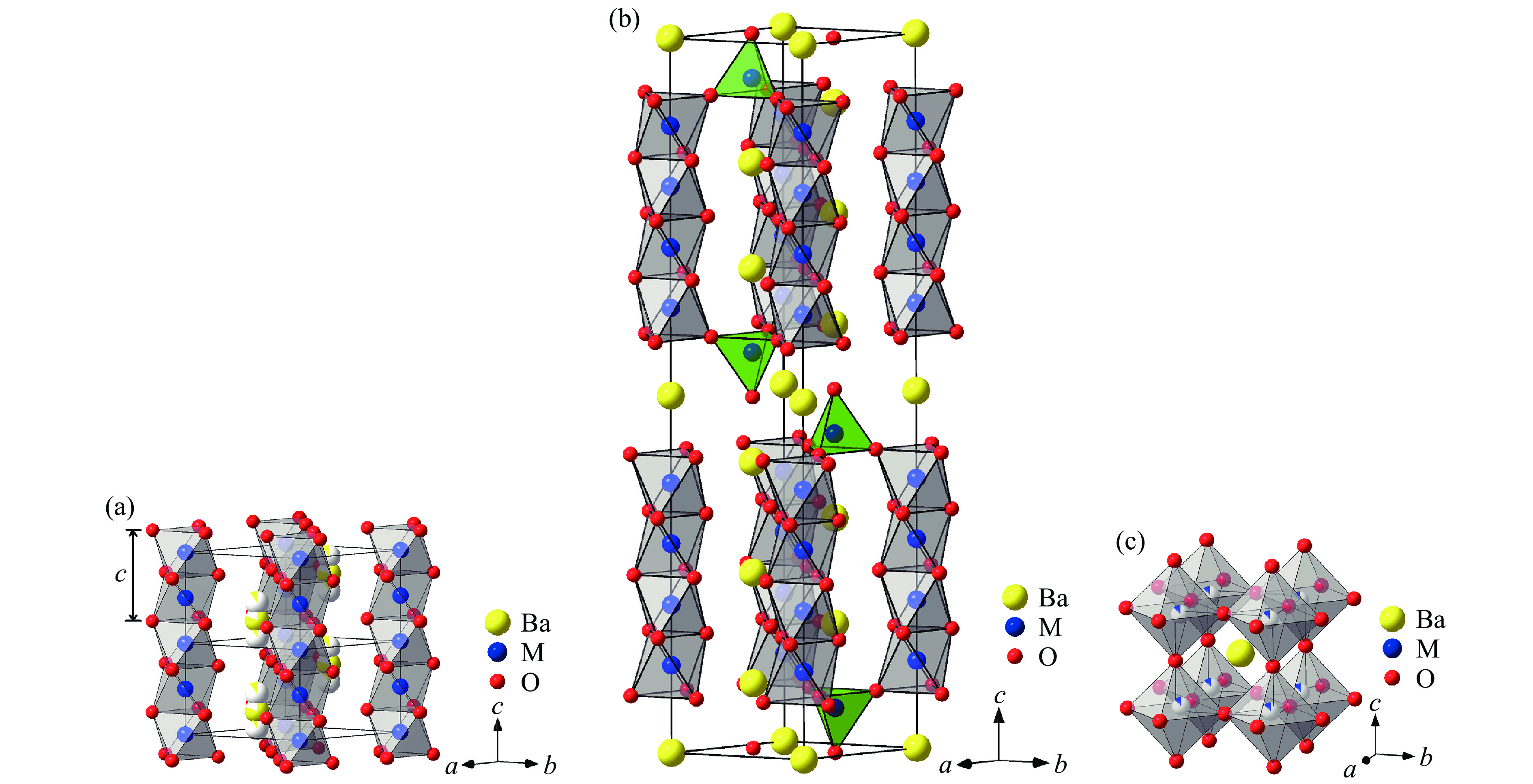
图 16 (a) 2H'-BaMO3、(b) 12H-BaMO3和(c) 3C'-BaMO3的晶体结构示意图
Figure 16. Schematic diagram of crystal structure of (a) 2H'-BaMO3, (b) 12H-BaMO3, and (c) 3C'-BaMO3
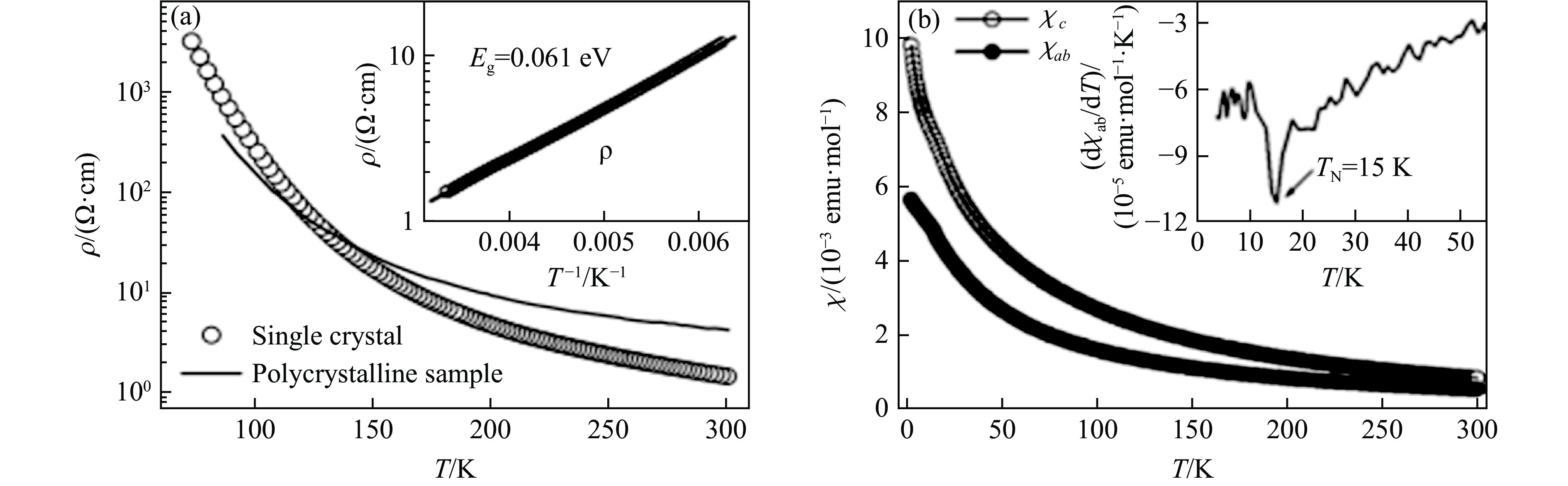
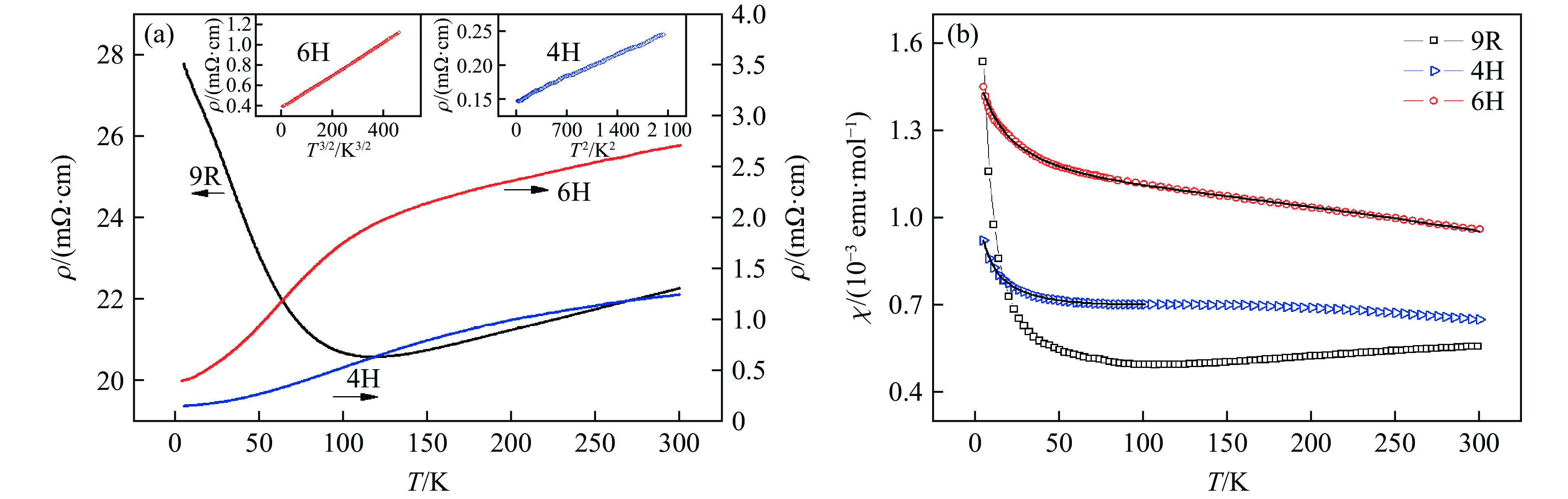
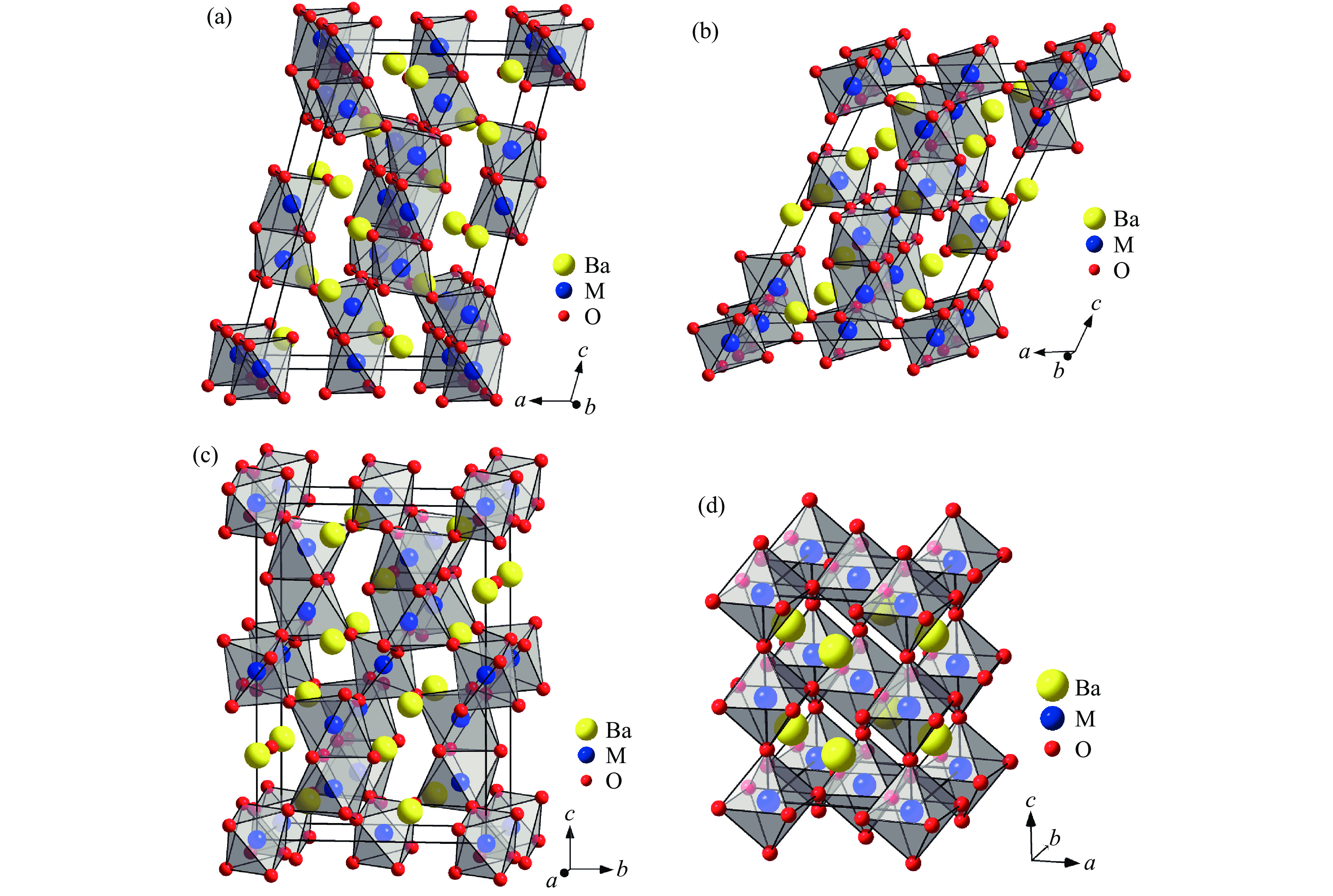
图 21 (a) 9M-BaMO3、(b) 5M-BaMO3、(c) 6M-BaMO3和(d) 四方钙钛矿BaMO3的晶体结构示意图
Figure 21. Schematic views of the crystal structure of (a) 9M-BaMO3, (b) 5M-BaMO3,(c) 6M-BaMO3, and (d) tetragonal perovskite BaMO3
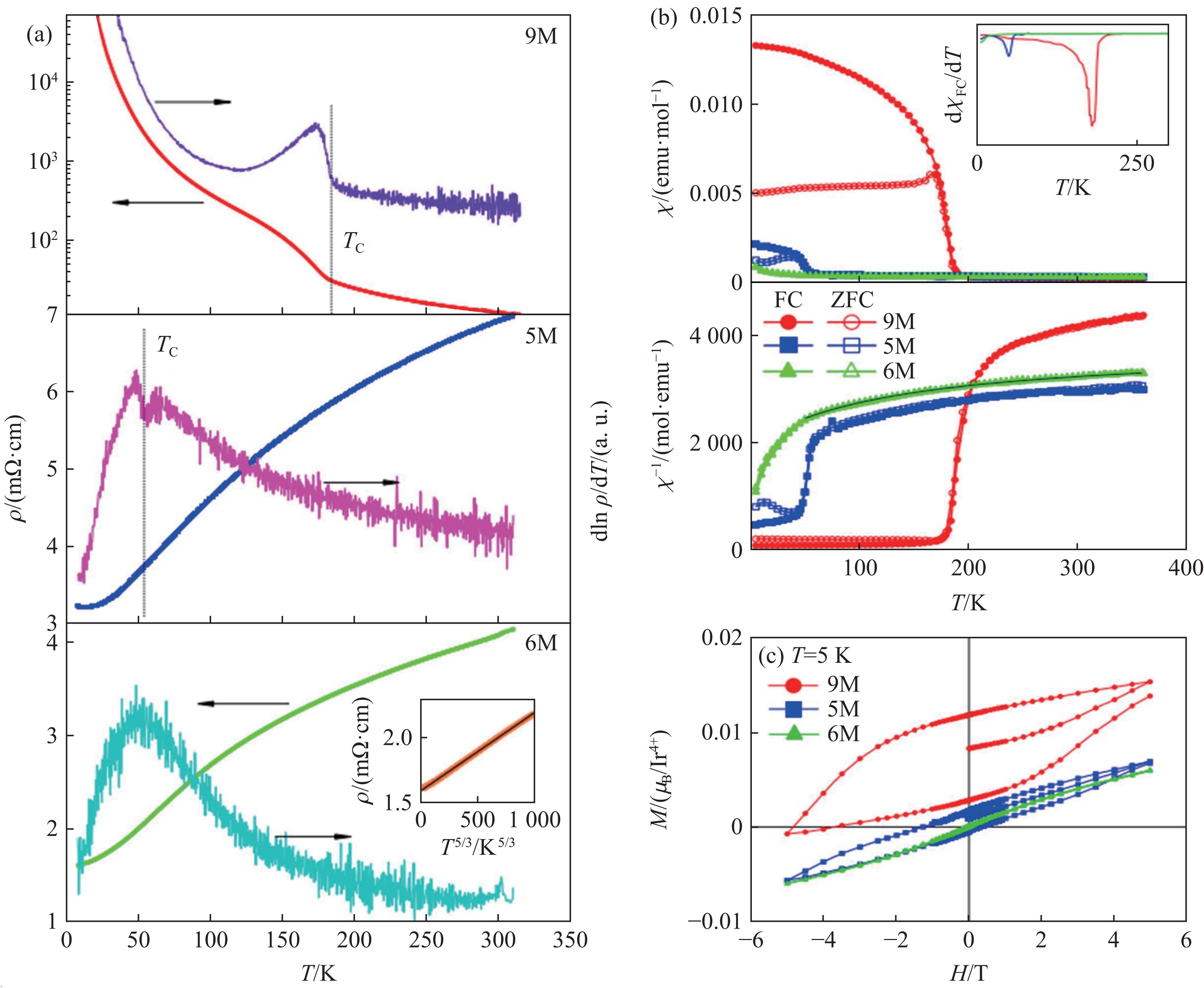
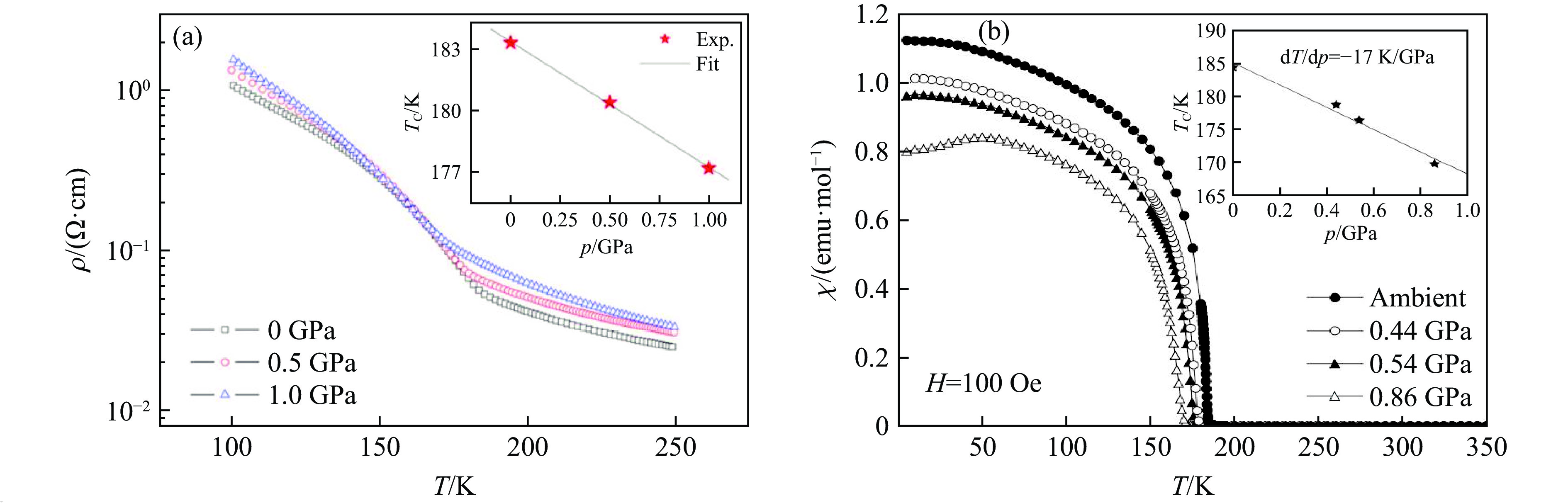
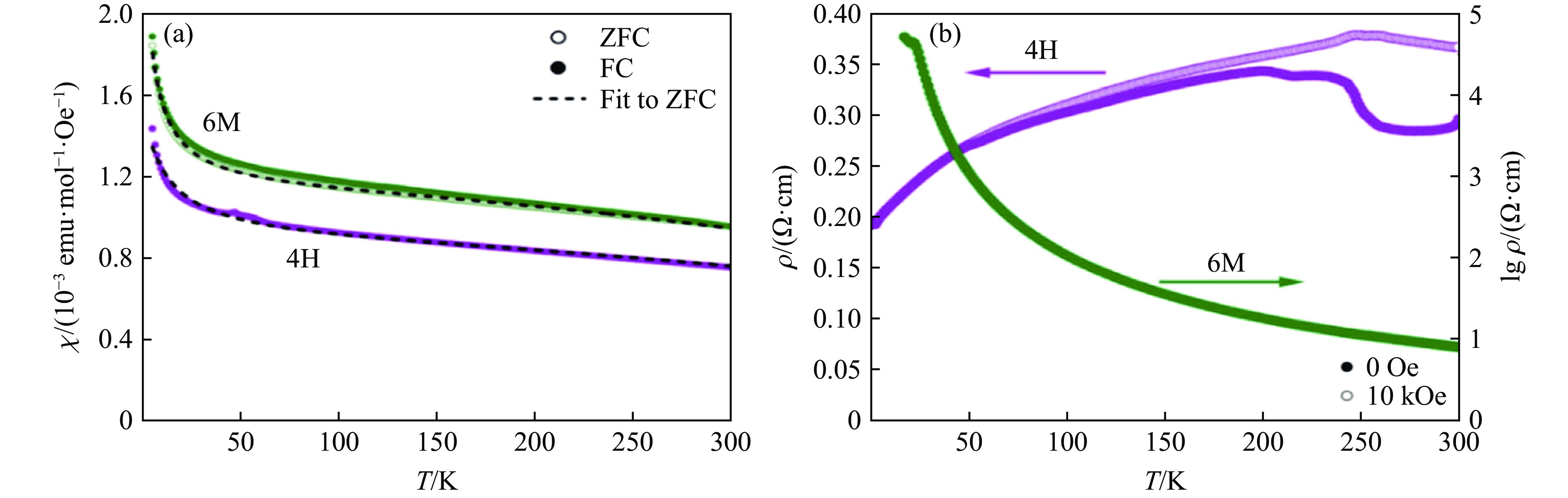
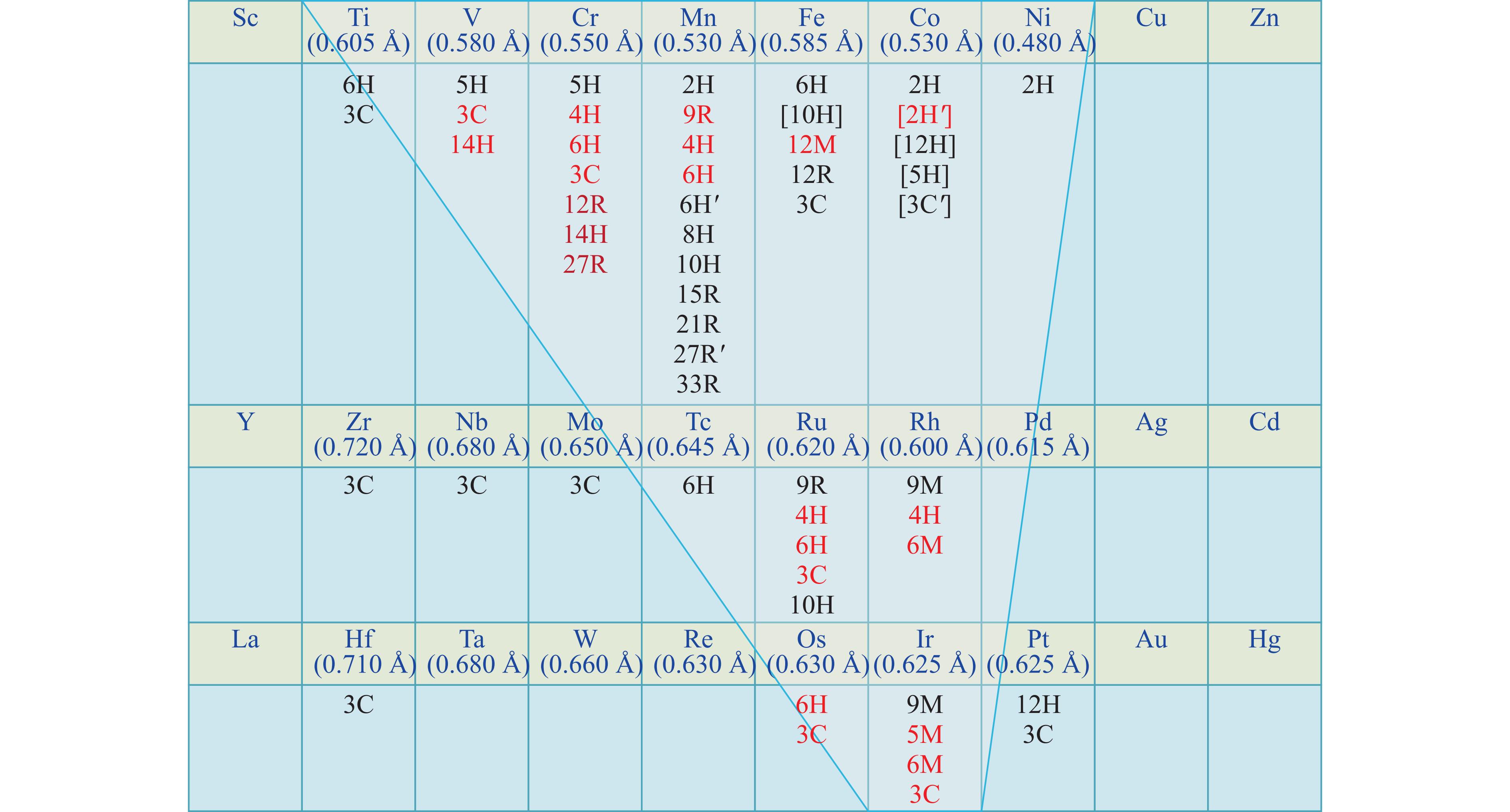
图 26 BaMO3(M为过渡金属离子)的多层堆积变体
Figure 26. Multi-layer stacked variants of BaMO3 (M is a transition metal)
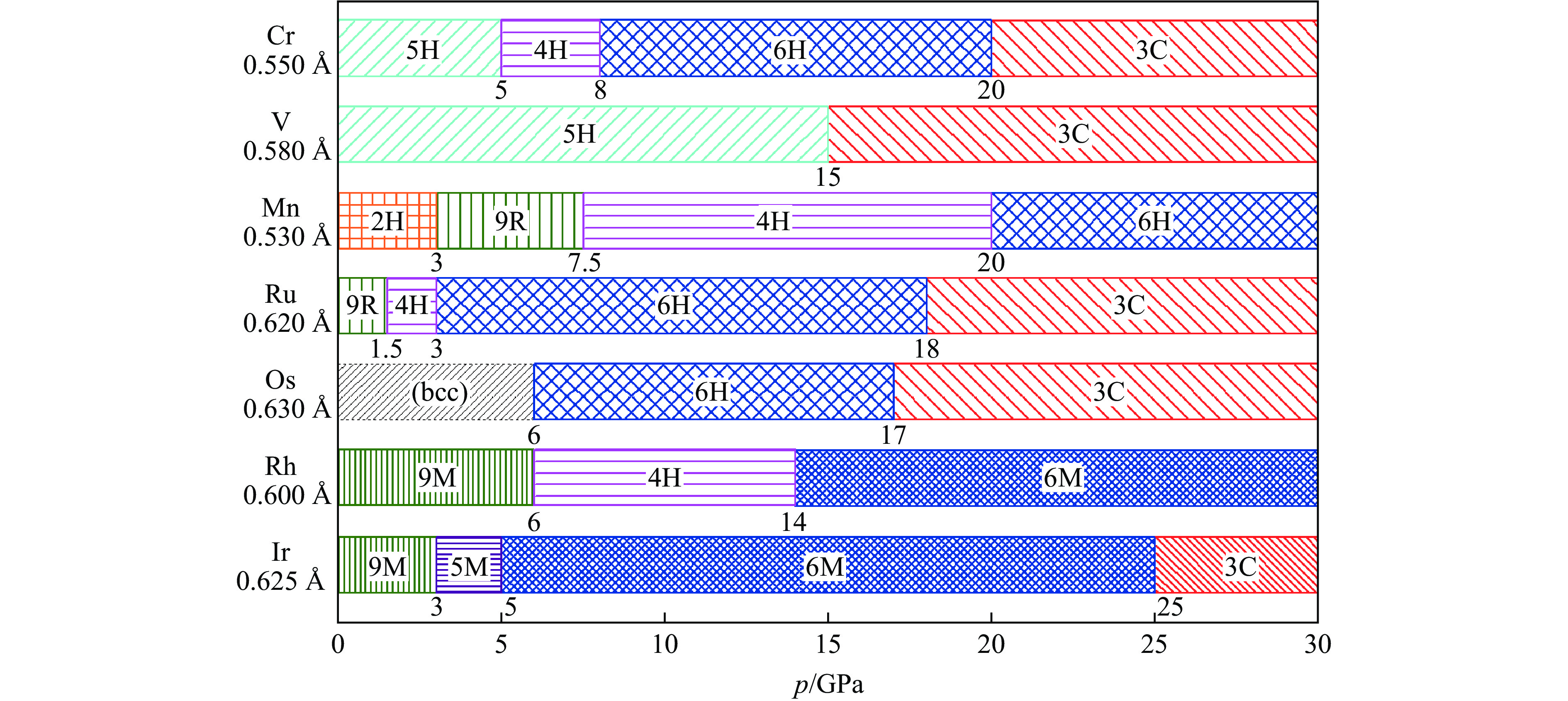
图 27 BaMO3(M = V, Cr, Mn, Ru, Os, Rh, Ir)在不同合成压力范围内的多层堆积变体
Figure 27. Multi-layer stacked variants of BaMO3 (M = V, Cr, Mn, Ru, Os, Rh, Ir) in different synthetic pressures
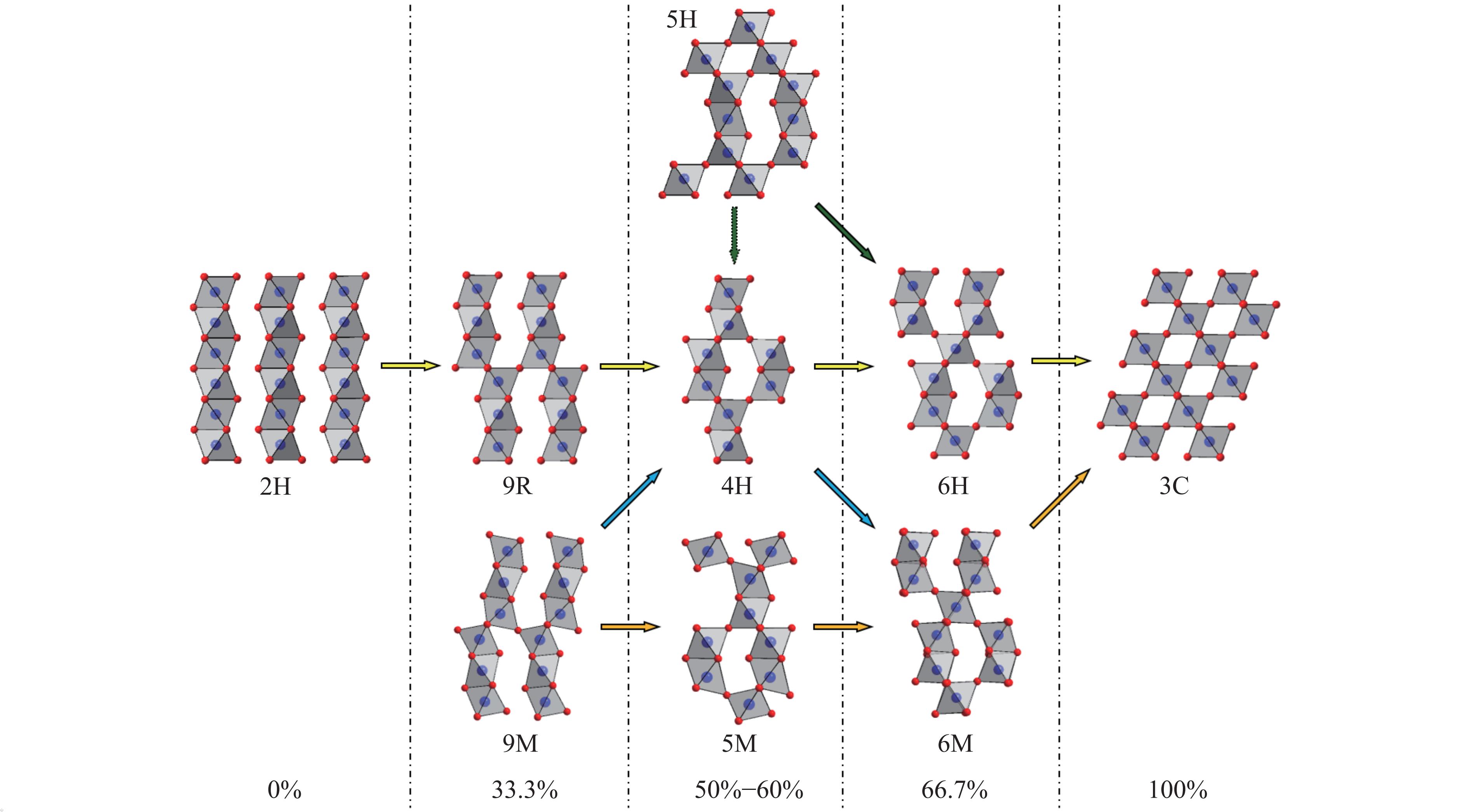
图 28 BaMO3中MO6八面体连接情况随合成压力的演化
Figure 28. Evolution of MO6 octahedron’s connectivity in BaMO3 with synthetic pressure
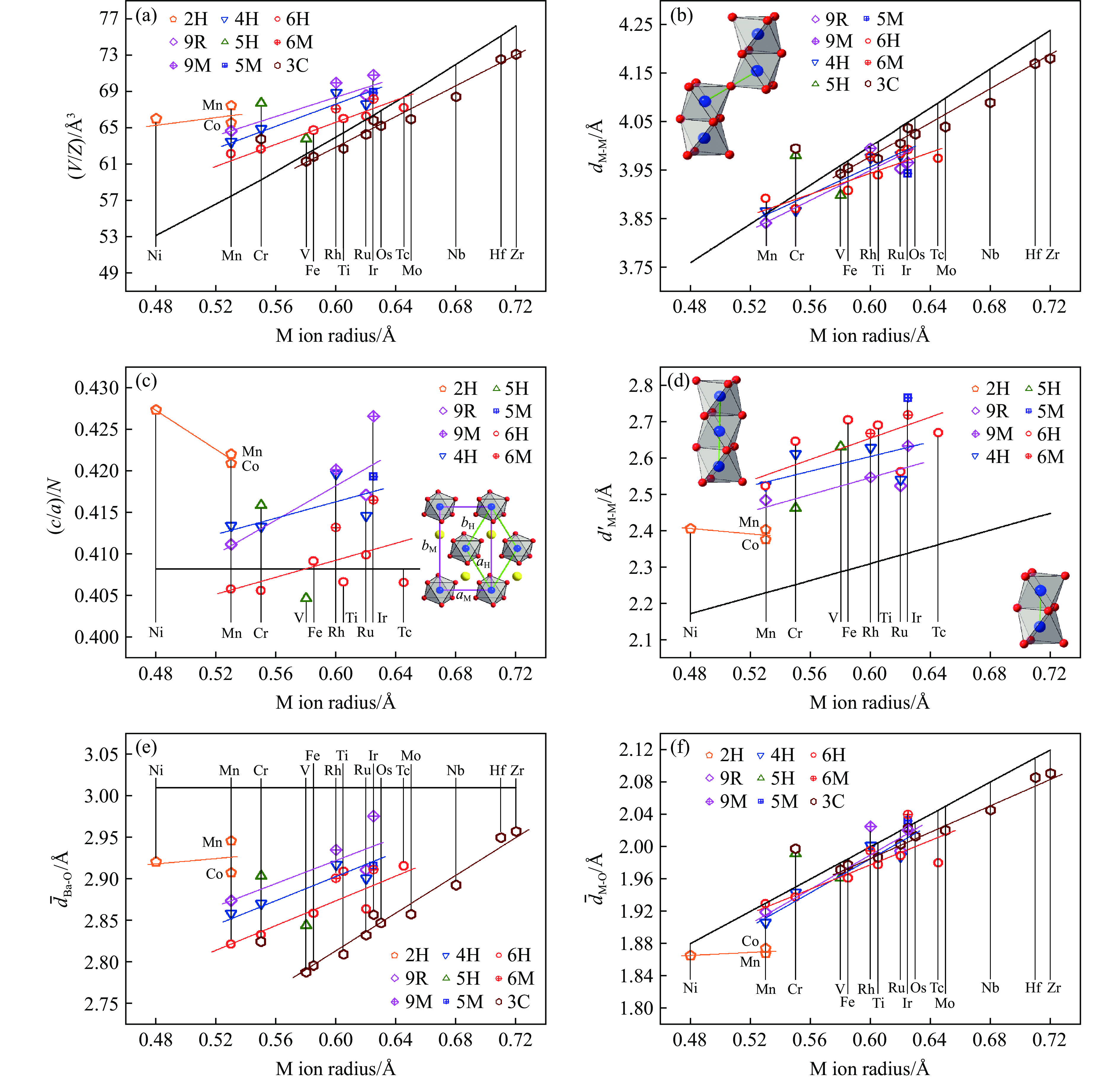
图 29 BaMO3的(a) 每个化学式的晶胞体积V/Z、(b) 近邻多聚体之间的M-M距离dM-M、(c) 轴比率(c/a)/N、(d) 多聚体内M-M之间的距离
${d'_{{\text{M-M}}}} $ ,(e) Ba-O之间的平均距离${\bar d_{{\text{Ba-O}}}}$ 和(f) M-O之间的平均距离${\bar d_{{\text{M-O}}}}$ 随M离子半径的变化关系Figure 29. Relations of (a) volume per chemical formula V/Z, (b) M-M distance between neighbour polymers dM-M,(c) axis ratio (c/a)/N, (d) M-M distance in one polymer
${d'_{{\text{M-M}}}} $ ; (e) average distance of Ba-O${\bar d_{{\text{Ba-O}}}}$ ; (f) average distance of M-O${\bar d_{{\text{M-O}}}}$ versus M ion radius of BaMO3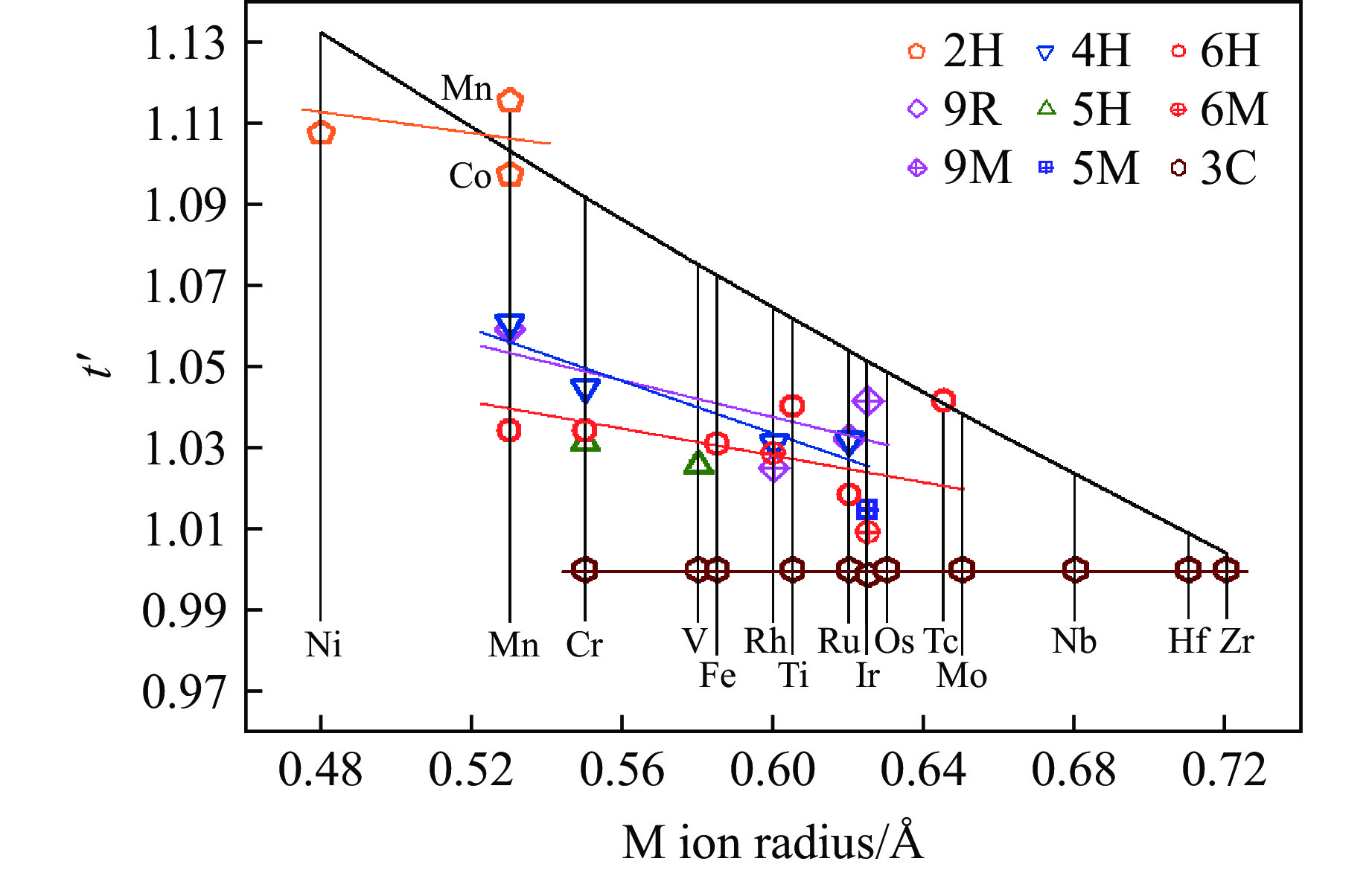
图 30 BaMO3的“容忍因子”t'随M离子半径的变化关系
Figure 30. Relationship of “tolerance factor” t' versus M ion radius of BaMO3
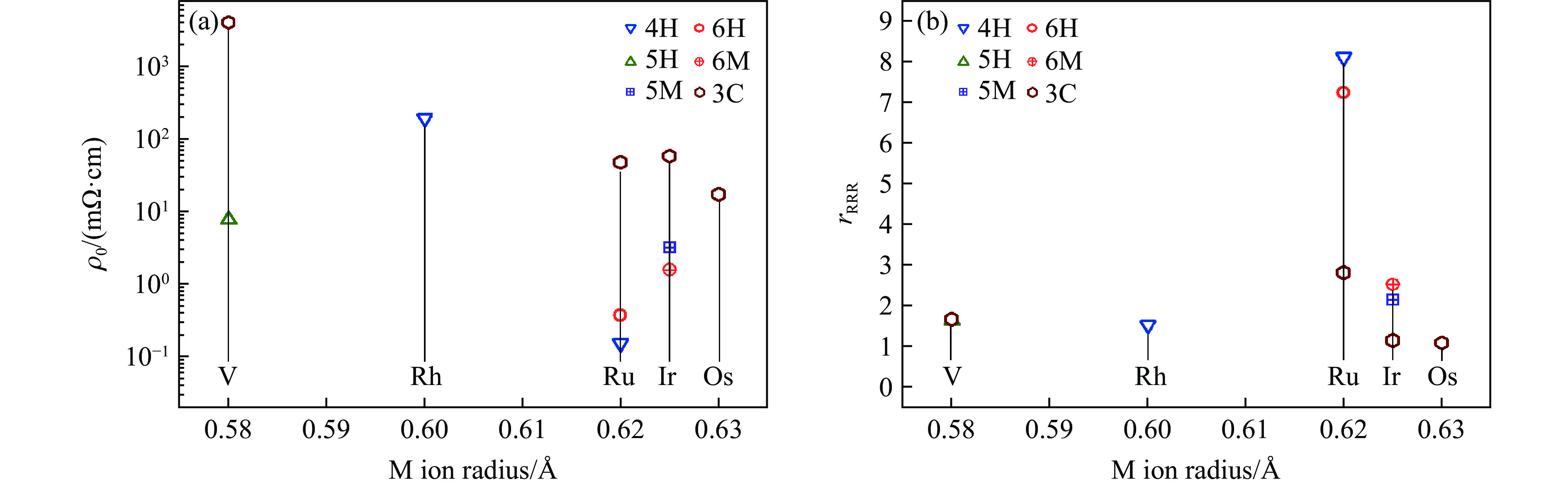
图 31 BaMO3的(a) 剩余电阻率ρ0和(b) 剩余电阻比率rRRR
Figure 31. (a) Residual resistivity ρ0 and (b) residual resistivity ratio rRRR of BaMO3
-
[1] GOLDSCHMIDT V M. Die gesetze der krystallochemie [J]. Naturwissenschaften, 1926, 14(21): 477–485. doi: 10.1007/BF01507527 [2] GLAZER A M. The classification of tilted octahedra in perovskites [J]. Acta Crystallographica Section B, 1972, 28(11): 3384–3392. doi: 10.1107/S0567740872007976 [3] SHANNON R D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides [J]. Acta Crystallographica Section A, 1976, 32(5): 751–767. doi: 10.1107/S0567739476001551 [4] NGUYEN L T, CAVA R J. Hexagonal perovskites as quantum materials [J]. Chemical Reviews, 2021, 121(5): 2935–2965. doi: 10.1021/acs.chemrev.0c00622 [5] AKIMOTO J, GOTOH Y, OSAWA Y. Refinement of hexagonal BaTiO3 [J]. Acta Crystallographica Section C, 1994, 50(2): 160–161. [6] HAYWARD S A, REDFERN S A T, STONE H J, et al. Phase transitions in BaTiO3: a high-pressure neutron diffraction study [J]. Zeitschrift für Kristallographie-Crystalline Materials, 2005, 220(8): 735–739. [7] LIU G, GREEDAN J E. Syntheses, structures, and characterization of 5-layer BaVO3− x (x = 0.2, 0.1, 0.0) [J]. Journal of Solid State Chemistry, 1994, 110(2): 274–289. doi: 10.1006/jssc.1994.1170 [8] NISHIMURA K, YAMADA I, OKA K, et al. High-pressure synthesis of BaVO3: a new cubic perovskite [J]. Journal of Physics and Chemistry of Solids, 2014, 75(6): 710–712. doi: 10.1016/j.jpcs.2014.02.001 [9] CHAMBERLAND B L, DANIELSON P S. Alkaline-earth vanadium (Ⅳ) oxides having the AVO3 composition [J]. Journal of Solid State Chemistry, 1971, 3(2): 243–247. doi: 10.1016/0022-4596(71)90035-1 [10] ARÉVALO-LÓPEZ A M, ATTFIELD J P. High-pressure BaCrO3 polytypes and the 5H-BaCrO2.8 phase [J]. Journal of Solid State Chemistry, 2015, 232: 236–240. doi: 10.1016/j.jssc.2015.09.029 [11] CHAMBERLAND B L. Crystal structure of the 4H BaCrO3 polytype [J]. Journal of Solid State Chemistry, 1982, 43(3): 309–313. doi: 10.1016/0022-4596(82)90245-6 [12] CHAMBERLAND B L. Crystal structure of the 6H BaCrO3 polytype [J]. Journal of Solid State Chemistry, 1983, 48(3): 318–322. doi: 10.1016/0022-4596(83)90088-9 [13] CHAMBERLAND B L. Preparation and crystallographic properties of barium chromate (Ⅳ) polytypes [J]. Inorganic Chemistry, 1969, 8(2): 286–290. doi: 10.1021/ic50072a021 [14] CHAMBERLAND B L, KATZ L. The structure of the fourteen-layer polytype of barium chromium trioxide, BaCrO3 [J]. Acta Crystallographica Section B, 1982, 38(1): 54–57. doi: 10.1107/S0567740882002039 [15] HARADEM P S, CHAMBERLAND B L, KATZ L. The structure of the 27-layer polytype of BaCrO3 [J]. Journal of Solid State Chemistry, 1980, 34(1): 59–64. doi: 10.1016/0022-4596(80)90403-X [16] ARÉVALO-LÓPEZ A M, REEVES S J, ATTFIELD J P. Ferrimagnetism in the high pressure 6H-perovskite BaCrO3 [J]. Zeitschrift für Anorganische und Allgemeine Chemie, 2014, 640(14): 2727–2729. [17] CUSSEN E J, BATTLE P D. Crystal and magnetic structures of 2H BaMnO3 [J]. Chemistry of Materials, 2000, 12(3): 831–838. doi: 10.1021/cm991144j [18] SYONO Y, AKIMOTO S I, KOHN K. Structure relations of hexagonal perovskite-like compounds ABX3 at high pressure [J]. Journal of the Physical Society of Japan, 1969, 26(4): 993–999. doi: 10.1143/JPSJ.26.993 [19] CHRISTENSEN A N, OLLIVIER G. Hydrothermal and high-pressure preparation of some BaMnO3 modifications and low-temperature magnetic properties of BaMnO3(2H) [J]. Journal of Solid State Chemistry, 1972, 4(1): 131–137. doi: 10.1016/0022-4596(72)90141-7 [20] BOULLAY P, HERVIEU M, LABBÉ P, et al. Single crystal and HREM study of the “Bi-Sr” stabilized BaMnO3 9R polytype [J]. Materials Research Bulletin, 1997, 32(1): 35–42. doi: 10.1016/S0025-5408(96)00169-9 [21] HARDY A. Structures cristallines de deux variétés allotropiques de manganite de baryum. Nouvelle structure ABO3 [J]. Acta Crystallographica, 1962, 15(3): 179–181. doi: 10.1107/S0365110X6200047X [22] QIN S J, CHIN Y Y, ZHOU B W, et al. High-pressure synthesis and magnetism of the 4H-BaMnO3 single crystal and its 6H-type polymorph [J]. Inorganic Chemistry, 2021, 60(21): 16308–16315. doi: 10.1021/acs.inorgchem.1c02155 [23] ADKIN J J, HAYWARD M A. BaMnO3− x revisited: a structural and magnetic study [J]. Chemistry of Materials, 2007, 19(4): 755–762. doi: 10.1021/cm062055r [24] POTOFF A D, CHAMBERLAND B L, KATZ L. A single crystal study of eight-layer barium managanese oxide, BaMnO3 [J]. Journal of Solid State Chemistry, 1973, 8(3): 234–237. doi: 10.1016/0022-4596(73)90090-X [25] PARRAS M, GONZÁLEZ-CALBET J M, ALONSO J, et al. Microstructural characterization of BaMnO3− y (0.08 ≤ y ≤ 0.12): evidence for a new polytype (21R) [J]. Journal of Solid State Chemistry, 1994, 113(1): 78–87. doi: 10.1006/jssc.1994.1344 [26] POOJITHA B, RATHORE A, KUMAR A, et al. Signatures of magnetostriction and spin-phonon coupling in magnetoelectric hexagonal 15R-BaMnO3 [J]. Physical Review B, 2020, 102(13): 134436. doi: 10.1103/PhysRevB.102.134436 [27] KORNETA O B, QI T F, GE M, et al. Correlated giant dielectric peaks and antiferromagnetic transitions near room temperature in pure and alkali-doped BaMnO3-δ [J]. Journal of Physics: Condensed Matter, 2011, 23(43): 435901. doi: 10.1088/0953-8984/23/43/435901 [28] GONZÁLEZ-CALBET J M, PARRAS M, ALONSO J, et al. Prediction of novel BaMnO3− y (0 < y < 0.1) perovskite related phases [J]. Journal of Solid State Chemistry, 1994, 111(1): 202–207. doi: 10.1006/jssc.1994.1218 [29] PARRAS M, VALLET-REGI M, GONZALEZ-CALBET J M, et al. A reassessment of Ba2Fe2O5 [J]. Materials Research Bulletin, 1987, 22(10): 1413–1419. doi: 10.1016/0025-5408(87)90306-0 [30] MORI K, KAMIYAMA T, KOBAYASHI H, et al. Structural evidence for the charge disproportionation of Fe4+ in BaFeO3−δ [J]. Journal of the Physical Society of Japan, 2003, 72(8): 2024–2028. doi: 10.1143/JPSJ.72.2024 [31] MORI K, KAMIYAMA T, KOBAYASHI H, et al. Mixed magnetic phase in 6H-type BaFeO3− δ [J]. Journal of Applied Crystallography, 2007, 40(Suppl 1): s501–s505. doi: 10.1107/S0021889807001653 [32] GÓMEZ M I, LUCOTTI G, DE MORÁN J A, et al. Ab initio structure solution of BaFeO2.8− δ, a new polytype in the system BaFeO y (2.5 ≤ y ≤ 3.0) prepared from the oxidative thermal decomposition of BaFe[(CN)5NO]·3H2O [J]. Journal of Solid State Chemistry, 2001, 160(1): 17–24. doi: 10.1006/jssc.2001.9119 [33] PARRAS M, VALLETREGI M, GONZALEZCALBET J M, et al. A structural study of 12H-BaFeO2.93 [J]. European Journal of Solid State and Inorganic Chemistry, 1989, 26(3): 299–312. [34] TAN Z H, ROMERO F D, SAITO T, et al. Charge disproportionation and interchange transitions in twelve-layer BaFeO3 [J]. Physical Review B, 2020, 102(5): 054404. doi: 10.1103/PhysRevB.102.054404 [35] HAYASHI N, YAMAMOTO T, KAGEYAMA H, et al. BaFeO3: a ferromagnetic iron oxide [J]. Angewandte Chemie International Edition, 2011, 50(52): 12547–12550. [36] MIZUMAKI M, YOSHII K, HAYASHI N, et al. Magnetocaloric effect of field-induced ferromagnet BaFeO3 [J]. Journal of Applied Physics, 2013, 114(7): 073901. doi: 10.1063/1.4818316 [37] LIU Y X, LIU Z H, LI Z, et al. Multiple magnetic transitions and electrical transport transformation of a BaFeO3 cubic perovskite single crystal [J]. Physical Review B, 2020, 101(14): 144421. doi: 10.1103/PhysRevB.101.144421 [38] STRAUSS S W, FANKUCHEN I, WARD R. Barium cobalt oxide of the perowskite type [J]. Journal of the American Chemical Society, 1951, 73(11): 5084–5086. doi: 10.1021/ja01155a019 [39] TAGUCHI H, TAKEDA Y, KANAMARU F, et al. Cobalt trioxide [J]. Acta Crystallographica Section B, 1977, 33(4): 1298–1299. doi: 10.1107/S0567740877005937 [40] WANG H D, YANG J H, DONG C H, et al. Crystal growth and characterization of the quasi-one-dimensional compound BaCoO3 [J]. Journal of Crystal Growth, 2015, 430: 52–54. doi: 10.1016/j.jcrysgro.2015.08.010 [41] SUGIYAMA J, NOZAKI H, BREWER J H, et al. Appearance of a two-dimensional antiferromagnetic order in quasi-one-dimensional cobalt oxides [J]. Physical Review B, 2005, 72(6): 064418. doi: 10.1103/PhysRevB.72.064418 [42] NOZAKI H, JANOSCHEK M, ROESSLI B, et al. Neutron diffraction and μSR study on the antiferromagnet BaCoO3 [J]. Physical Review B, 2007, 76(1): 014402. doi: 10.1103/PhysRevB.76.014402 [43] BOTTA P M, PARDO V, BALDOMIR D, et al. Dynamic magnetic behavior of BaCoO3 quasi-one-dimensional perovskite [J]. Physical Review B, 2006, 74(21): 214415. doi: 10.1103/PhysRevB.74.214415 [44] WANG H Z, XU X H, NI D R, et al. Impersonating a superconductor: high-pressure BaCoO3, an insulating ferromagnet [J]. Journal of the American Chemical Society, 2023, 145(39): 21203-21206. [45] JACOBSON A J, HUTCHISON J L. An investigation of the structure of 12H BaCoO2.6 by electron microscopy and powder neutron diffraction [J]. Journal of Solid State Chemistry, 1980, 35(3): 334–340. doi: 10.1016/0022-4596(80)90530-7 [46] PARRAS M, VARELA A, SEEHOFER H, et al. HREM study of the BaCoO3− y system: evidence for a new 5H phase [J]. Journal of Solid State Chemistry, 1995, 120(2): 327–331. doi: 10.1006/jssc.1995.1416 [47] MENTRÉ O, IORGULESCU M, HUVÉ M, et al. BaCoO2.22: the most oxygen-deficient certified cubic perovskite [J]. Dalton Transactions, 2015, 44(23): 10728–10737. [48] LANDER J J. The crystal structures of NiO·BaO3, NiO·BaO, BaNiO3 and intermediate phases with composition near Ba2Ni2O5, with a note on NiO [J]. Acta Crystallographica, 1951, 4(2): 148–156. doi: 10.1107/S0365110X51000441 [49] TAKEDA Y, SHIMADA M, KANAMARU F, et al. Preparation and magnetic property of BaNiO3 single crystals [J]. Chemistry Letters, 1974, 3(2): 107–108. doi: 10.1246/cl.1974.107 [50] DONOHUE P C, KATZ L, WARD R. The crystal structure of barium ruthenium oxide and related compounds [J]. Inorganic Chemistry, 1965, 4(3): 306–310. doi: 10.1021/ic50025a010 [51] RAO M V R, SATHE V G, SORNADURAI D, et al. Electronic structure of ARuO3 (A = Ca, Sr and Ba) compounds [J]. Journal of Physics and Chemistry of Solids, 2001, 62(4): 797–806. doi: 10.1016/S0022-3697(00)00262-6 [52] HONG S T, SLEIGHT A W. Crystal structure of 4H BaRuO3: high pressure phase prepared at ambient pressure [J]. Journal of Solid State Chemistry, 1997, 128(2): 251–255. [53] RIJSSENBEEK J T, JIN R, ZADOROZHNY Y, et al. Electrical and magnetic properties of the two crystallographic forms of BaRuO3 [J]. Physical Review B, 1999, 59(7): 4561–4564. doi: 10.1103/PhysRevB.59.4561 [54] ZHAO J G, YANG L X, YU Y, et al. Structural and physical properties of the 6H BaRuO3 polymorph synthesized under high pressure [J]. Journal of Solid State Chemistry, 2007, 180(10): 2816–2823. doi: 10.1016/j.jssc.2007.07.031 [55] JIN C Q, ZHOU J S, GOODENOUGH J B, et al. High-pressure synthesis of the cubic perovskite BaRuO3 and evolution of ferromagnetism in ARuO3 (A = Ca, Sr, Ba) ruthenates [J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(20): 7115–7119. [56] OGAWA T, SATO H. New ternary barium ruthenates: 10H-type BaRuO3 and Ba2Ru7O18 [J]. Journal of Alloys and Compounds, 2004, 383(1/2): 313–318. [57] ZHOU J S, MATSUBAYASHI K, UWATOKO Y, et al. Critical behavior of the ferromagnetic perovskite BaRuO3 [J]. Physical Review Letters, 2008, 101(7): 077206. doi: 10.1103/PhysRevLett.101.077206 [58] NEUMEIER J J, CORNELIUS A L, SCHILLING J S. Influence of pressure on the ferromagnetic transition temperature of SrRuO3 [J]. Physica B: Condensed Matter, 1994, 198(4): 324–328. [59] SIEGRIST T, CHAMBERLAND B L. The crystal structure of BaIrO3 [J]. Journal of the Less Common Metals, 1991, 170(1): 93–99. doi: 10.1016/0022-5088(91)90054-8 [60] CHENG J G, ALONSO J A, SUARD E, et al. A new perovskite polytype in the high-pressure sequence of BaIrO3 [J]. Journal of the American Chemical Society, 2009, 131(21): 7461–7469. [61] ZHAO J G, YANG L X, YU Y, et al. Physical properties of the 5M BaIrO3: a new weak ferromagnetic iridate synthesized under high pressure [J]. Solid State Communications, 2010, 150(1): 36–39. [62] ZHAO J G, YANG L X, YU Y, et al. Structural and physical properties of the 6M BaIrO3: a new metallic iridate synthesized under high pressure [J]. Inorganic Chemistry, 2009, 48(10): 4290–4294. [63] CHENG J G, ISHII T, KOJITANI H, et al. High-pressure synthesis of the BaIrO3 perovskite: a Pauli paramagnetic metal with a Fermi liquid ground state [J]. Physical Review B, 2013, 88(20): 205114. doi: 10.1103/PhysRevB.88.205114 [64] CHENG J G, ZHOU J S, ALONSO J A, et al. Transition from a weak ferromagnetic insulator to an exchange-enhanced paramagnetic metal in the BaIrO3 polytypes [J]. Physical Review B, 2009, 80(10): 104430. doi: 10.1103/PhysRevB.80.104430 [65] CAO G, CROW J E, GUERTIN R P, et al. Charge density wave formation accompanying ferromagnetic ordering in quasi-one-dimensional BaIrO3 [J]. Solid State Communications, 2000, 113(11): 657–662. doi: 10.1016/S0038-1098(99)00532-3 [66] POWELL A V, BATTLE P D. The electronic properties of non-stoichiometric barium iridate, BaIrO3− δ [J]. Journal of Alloys and Compounds, 1993, 191(2): 313–318. doi: 10.1016/0925-8388(93)90085-2 [67] ZHAO J G, YANG L X, MYDEEN K, et al. Effects of pressure on electrical property of BaIrO3 [J]. Solid State Communications, 2008, 148(9/10): 361–364. [68] KIDA T, SENDA A, YOSHII S, et al. Pressure effect on magnetic properties of a weak ferromagnet BaIrO3 [J]. Journal of Physics: Conference Series, 2010, 200(1): 012084. doi: 10.1088/1742-6596/200/1/012084 [69] SIEGRIST T, LARSON E M, CHAMBERLAND B L. Structural studies of high-pressure Ba-Rh-O phases [J]. Journal of Alloys and Compounds, 1994, 210(1/2): 13–17. [70] CHAMBERLAND B L, ANDERSON J B. The preparation and crystal structure of a BaRhO3 polytype [J]. Journal of Solid State Chemistry, 1981, 39(1): 114–119. doi: 10.1016/0022-4596(81)90309-1 [71] INJAC S D A, XU Y H, ROMERO F D, et al. Pauli-paramagnetic and metallic properties of high pressure polymorphs of BaRhO3 oxides containing Rh2O9 dimers [J]. Dalton Transactions, 2021, 50(13): 4673–4679. doi: 10.1039/D1DT00502B [72] MEGAW H D. Crystal structure of double oxides of the perovskite type [J]. Proceedings of the Physical Society, 1946, 58(2): 133–152. doi: 10.1088/0959-5309/58/2/301 [73] KOPNIN E M, ISTOMIN S Y, D’YACHENKO O G, et al. Synthesis, structure, and resistivity properties of K1− xBa xNbO3 (0.2 ≤ x ≤ 0.5) and K0.5Sr0.5NbO3 [J]. Materials Research Bulletin, 1995, 30(11): 1379–1386. doi: 10.1016/0025-5408(95)00117-4 [74] CASAIS M T, ALONSO J A, RASINES I, et al. Preparation, neutron structural study and characterization of BaNbO3: a Pauli-like metallic perovskite [J]. Materials Research Bulletin, 1995, 30(2): 201–208. [75] BRIXNER L H. X-ray study and electrical properties of system Ba xSr1− xMoO3 [J]. Journal of Inorganic and Nuclear Chemistry, 1960, 14(3/4): 225–230. [76] SCHOLDER R, RÄDE D, SCHWARZ H. Über zirkonate, hafnate und thorate von barium, strontium, lithium und natrium [J]. Zeitschrift für Anorganische und Allgemeine Chemie, 1968, 362(3/4): 149–168. [77] MULLER O, WHITE W B, ROY R. Crystal chemistry of some technetium-containing oxides [J]. Journal of Inorganic and Nuclear Chemistry, 1964, 26(12): 2075–2086. doi: 10.1016/0022-1902(64)80152-4 [78] SARKOZY R F, CHAMBERLAND B L. The preparation of several new ternary oxides of osmium [J]. Materials Research Bulletin, 1973, 8(12): 1351–1359. doi: 10.1016/0025-5408(73)90019-6 [79] CHAMBERLAND B L. Solid state preparations and reactions of ternary alkaline-earth osmium oxides [J]. Materials Research Bulletin, 1978, 13(12): 1273–1280. doi: 10.1016/0025-5408(78)90117-4 [80] SHI Y G, GUO Y F, SHIRAKO Y, et al. High-pressure synthesis of 5d cubic perovskite BaOsO3 at 17 GPa: ferromagnetic evolution over 3d to 5d series [J]. Journal of the American Chemical Society, 2013, 135(44): 16507–16516. doi: 10.1021/ja4074408 [81] GALLAGHER P K, JOHNSON JR D W, VOGEL E M, et al. Synthesis and structure of BaPtO3 [J]. Journal of Solid State Chemistry, 1977, 21(4): 277–282. doi: 10.1016/0022-4596(77)90126-8 [82] CASAPU M, GRUNWALDT J D, MACIEJEWSKI M, et al. Enhancement of activity and self-reactivation of NSR-catalysts by temporary formation of BaPtO3-perovskite [J]. Catalysis Letters, 2008, 120(1/2): 1–7. [83] YAMAMOTO T, SHITARA K, KITAGAWA S, et al. Selective hydride occupation in BaVO3− xH x (0.3 ≤ x ≤ 0.8) with face and corner-shared octahedra [J]. Chemistry of Materials, 2018, 30(5): 1566–1574. doi: 10.1021/acs.chemmater.7b04571 [84] YUSA H, SATA N, OHISHI Y. Rhombohedral (9R) and hexagonal (6H) perovskites in barium silicates under high pressure [J]. American Mineralogist, 2007, 92(4): 648–654. [85] HIRAMATSU H, YUSA H, IGARASHI R, et al. An exceptionally narrow band-gap (~4 eV) silicate predicted in the cubic perovskite structure: BaSiO3 [J]. Inorganic Chemistry, 2017, 56(17): 10535–10542. doi: 10.1021/acs.inorgchem.7b01510 [86] 谢亚飞, 姜昌国, 罗兴丽, 等. 6H型六方钙钛矿相BaGeO3 的高温高压合成 [J]. 高压物理学报, 2021, 35(5): 051201. doi: 10.11858/gywlxb.20210761 XIE Y F, JIANG C G, LUO X L, et al. Synthesis of 6H-type hexagonal perovskite phase of BaGeO3 at high temperature and high pressure [J]. Chinese Journal of High Pressure Physics, 2021, 35(5): 051201. doi: 10.11858/gywlxb.20210761 [87] LONGO J M, KAFALAS J A. Pressure-induced structural changes in the system Ba1− xSr xRuO3 [J]. Materials Research Bulletin, 1968, 3(8): 687–692. [88] ZHAO J G, YANG L X, YU Y, et al. High-pressure synthesis of orthorhombic SrIrO3 perovskite and its positive magnetoresistance [J]. Journal of Applied Physics, 2008, 103(10): 103706. doi: 10.1063/1.2908879 [89] CAO G, BOLIVAR J, MCCALL S, et al. Weak ferromagnetism, metal-to-nonmetal transition, and negative differential resistivity in single-crystal Sr2IrO4 [J]. Physical Review B, 1998, 57(18): R11039–R11042. doi: 10.1103/PhysRevB.57.R11039 [90] CAO G, XIN Y, ALEXANDER C S, et al. Anomalous magnetic and transport behavior in the magnetic insulator Sr3Ir2O7 [J]. Physical Review B, 2002, 66(21): 214412. doi: 10.1103/PhysRevB.66.214412 -
计量
- 文章访问数: 1882
- HTML全文浏览数: 1882
- PDF下载数: 16
- 施引文献: 0