
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
光致变色是指一类化合物在受到一定波长的光激发后, 可进行单向或双向的可逆化学反应, 如光致异构化或光物理效应, 其获得的产物由于结构改变导致吸收光谱发生明显变化的现象[1]. 有机光致变色化合物(分子光开关)也可在光照作用下可逆地改变颜色, 这一特质使其在现代高科技分子材料领域具有非常巨大的潜在应用价值, 从纳米技术到药理学方面都有广泛的应用前景, 因此引起了科学家的广泛关注[2–4]. 利用光开关可以促进光响应性药物载体、生物黏附介体、聚合物–酶开关、电子器件和分子机器的发展[5,6]. 通过光异构化反应, 光致变色分子可以增强材料的光物理、机械、电子或热学性质, 因此研究光致变色机理对材料科学领域的发展具有非常重要的意义[7,8]. 经过150多年的发展, 光致变色材料的发展已成为一个成熟的领域. 通过化学合成、时间分辨光谱以及理论研究的结合[9,10] , 加强了人们对光致变色机制的理解, 并为合理设计这类材料提供了思路[2].
光致变色逆反应分为热驱动(T型光致变色)或光化学驱动(P型光致变色)[11]. 但由于T型光致变色染料会随着热效应出现褪色, 因此其不适用于某些关键的高科技应用. 而光致变色P型衍生物在光学和光电子学中表现出更多的应用价值[12–14] , 包括光开关和逻辑门、数据存储中的光存储、望远镜的焦平面镀膜、全息影像、光纳米印刷术、波导中的光学调制等. 在众多的光致变色复合物中, 腙光开关代表着一类非常重要的P型光开关, 主要是基于其具有非常高的热稳定性(半衰期可达5300年), 易合成、易修饰、高的光稳定性及双稳态的荧光开关反应[15,16]. 许多腙类衍生物及其金属配合物具有多种生物效应, 因此, 腙类化合物具有很广泛的应用价值, 特别是在药学和医学领域[17,18]. 腙类化合物在多种超分子化学领域得到了广泛的应用, 如光致变色分子开关、光敏及热敏超分子排列以及作为比色或及荧光化学传感器等[19–23]. 在分子开关领域, 包括苯、奈、吲哚(靛红)等在内的光致发色团体系已被开发使用过[24] , 但腙类化合物分子开关的开发还相对较少. 光致变色衍生物种类很多, 包括常见的二芳基乙烯、俘精酸酐、螺吡喃[25] 、螺恶嗪、以及偶氮苯, 但是只有前两者才是典型的P型光致变色染料, 通常来讲, 通过简单的结构修饰就可以实现T型到P型的转变. 例如, Temps等[26]和Woolley等[27]研究了偶氮苯光开关在生物应用中的适用性, 大多数基于偶氮苯的光开关都需要使用紫外光照射来完成光异构化, 紫外光对生物活性有很大损害, 会限制它们在生物系统中的应用. 因此, 开发易于合成的可见光-可见光(可见光-近红外光)的P型光致变色染料仍然是当前光化学中的一个独特挑战[28–30].
具有分子开关特性的有机材料通常使用光作为产生开/关状态的外部刺激, 可选作光致变色分子开关的发色团通常需要满足以下几个条件: 一是对光照有响应, 能够在一定激光照射下完成光致异构化反应; 二是可在不同的最大峰值光照下完成可逆光致异构化反应. 吸收的光能转化为动能时会引起分子几何形状的改变对应于开或关的状态. 处于激发态的分子相对于基态来讲具有不同的电子分布和几何结构. 通过分子的不饱和发色团如C=C, C=N以及N=N的旋转可以实现光能到化学能的转换. 将光能转化为分子的机械能引起的分子构型的改变, 使得发色团可在两个或多个异构体之间进行转换, 这就相当于分子开或关的作用[31]. 最近, Cigán等[32]设计合成了一种靛红N2-二苯腙化合物, 它为新型可见光-可见光响应的分子开关提供基本骨架. 实验发现这类新的靛红衍生物E, Z构型具有不同的吸收峰值, 且它们的吸收光波段属于电磁光谱的可见区域, 同时研究表明它可以围绕C=N双键发生旋转异构化, 从而使之具有开/关功能的光致变色能力. 因其具有顺反异构化可见光激发特征, 被认为是非常适合做分子开关的一类化合物, 它在生物技术等领域具有极其广泛的应用价值. 其衍生物的易合成性、对外界刺激的敏感性都使得靛红双氮二苯腙化合物在分子开关及光量测定方面有极其重要的应用价值.
目前, 已有研究组对该新型分子开关及其衍生物的功能特性进行了系列研究报道. Cigan等[33]研究发现将强阴离子加入到苯腙溶液可以加强靛红NH基团的去质子化, 生成具有明显最大吸收峰差值的可见光-可见光响应的化合物, 表现出与负离子之间较强的相互作用, 这一特征增强了它作为分子开关的功能性, 使之成为单分子可实现的四态分子开关. Tisovský等[34] 研究了自由金属阳离子对其衍生物靛红二芳基腙化合物光致开关作用的影响, 实现了程序可控的可逆改变. 随后, 该研究组还在实验上合成了系列基于靛蓝的光致变色性能和靛红N2-芳基腙衍生物[32,33] , 并研究了其与各种二价金属阳离子的芳基腙配合物的合成及其生物活性[35] , 在这些复杂结构中, 金属离子阻止配体围绕C=N键旋转, 因此配体失去开/关功能. 后期, 该研究组还陆续报道了靛红联吡啶腙衍生物作为分子开关的特性, 及其N阴离子与联吡啶片段的合成与Zn配位的化学物性质等, 证明了其具有分子开关光学可逆反应特性[36].
在过去十几年, 腙类分子开关已经被集成应用到多种超分子光化学领域, 靛红双氮二苯腙化合物作为腙类分子的衍生物, 可以将吸收光波调整到可见光波段, 它的双稳态对光刺激有更高的敏感性, 因此在超分子光化学领域的集成应用也更容易实现. 这种双稳态分子开关法已被应用于调制重要的生物过程. 包括核酸的构建极其功能化、转运、蛋白折叠、酶活性、蛋白-配位反应、肽构建及其功能化、膜传输、受体调制及其信号传输[28,37]. 例如, Tochitsky等[38]最近介绍了一种新的恢复视觉的方法, 将可对光响应的分子开关结合在视觉神经蛋白上, 通过光致异构化的开关作用刺激视紫红蛋白折叠变形, 从而达到基因表达的作用. 通过二芳基乙烯合成的胆固醇两亲性分子开关作用, 还能控制囊泡及超分子有机凝胶的形成[39]. 值得一提的是, 基于过度拥挤烯烃及含N羟基亚胺的分子体系被用来合成了第一代分子马达[40] , 通过控制分子体系的旋转方向, 使得分子体系可以在光照驱动下持续不断地朝着一个方向旋转, 就可以完成分子马达的功能. 截至目前为止, Kistemaker等[41,42]开发的人造分子马达也已经更新到第三代.
尽管实验上已成功合成这类新型可见光波段响应的腙类发色团, 但是其内部详细的异构化机理并不清楚, 除熟知的围绕中心C=N键的旋转异构机制外, 内酰胺-内酰亚靛红互变异构体的平衡是否会触发新的光开关异构机制, 在电子退激发过程中分子体系是否会发生新奇的物理现象, 这些问题的回答都需要辅助详细的非绝热动力学研究. 因此, 本文在半经验OM2/MRCI水平上采用非绝热轨道面跳跃分子动力学方法, 系统揭示靛红N2-二苯腙的E-Z详细异构化反应机理. 此外, 超快时间分辨瞬态荧光光谱技术是研究皮秒和飞秒时间尺度的激发态动力学的有效手段, 它可以像超快摄像机一样, 直接观察处于化学反应过程中的原子与分子的转变状态, 因为其探测的信号均来自于电子激发态, 使得探究物理化学反应过程变得更加直接可靠. 分子体系在受到外界激光照射后, 吸收光子后会跃迁到激发态. 随后, 体系从激发态通过非辐射跃迁通道退激发回到基态, 并在这个过程中释放出具有一定波长和强度的荧光光子. 通过光学设备检测并分析荧光光子的强度及波长信息, 就可以了解光化学反应机理与路径. 在实验上, 通过超快时间分辨瞬态荧光光谱技术来进行直接探测已成为可能, 通过瞬间获得体系的状态, 从而研究体系在极短时间内的光物理、光化学和光生物反应. 本文也通过理论方法计算时间分辨荧光辐射谱, 以帮助深入理解超快动力学化学反应机制, 并探测新奇的反应现象.
本文结构安排如下: 第2节主要介绍非绝热动力学的研究方法及时间分辨荧光辐射谱的基本理论; 第3节系统揭示分子开关的两种光致异构化机制及荧光辐射谱预测的极快的荧光猝灭现象; 第4节是对分子开关的总结与展望.
-
本文采用基于Tully的最小轨迹面跳跃非绝热动力学模拟方法对新型分子开关的光致异构化反应进行计算, 利用半经验的正交化校正OM2方法结合GUGA-MRCI组态获得参考能、梯度和非绝热耦合量[43]. 所有半经验计算均采用MNDO99程序[43] 开发版本中实施的OM2/MRCI方法进行. 这种方法可以很好地平衡计算成本和精度, 并已成功地用于许多光诱导的光化学光物理反应过程的计算. 所需的能量、梯度、非绝热耦合因子、几何优化及动力学模拟都是基于这个方法得到的计算结果. 特别是, 通过在完全活性空间自洽场(CASSCF)理论框架内进行量子化学计算和非绝热分子动力学(NAMD)模拟, 可以很好地模拟出完整的光致异构化过程[44]. 本文中模拟所使用的多参考组态选取三种, 包括闭壳层基态构型以及从最高占据分子轨道(HOMO)到最低未占分子轨道(LUMO)的单激发和双激发[45]. 计算中选取的MRCI活性空间组态为14个电子占据13个轨道, 模拟证明该活性空间的选取不仅在构型优化与动力学中是非常合适的, 同时在计算锥形交叉结构(CI)时也是非常可靠的. 计算中使用了PIPIO的方法, 采用0.4的阈值来追踪π轨道并且确保π轨道始终在活性空间范围内. 其中, 基态S0与第一激发态S1的最优结构计算采用上面提及的OM2/MRCI方法, 而最优CI结构的获取则使用拉格朗日-牛顿算法来获取.
半经验OM2/MRCI是一种计算效率较高的多参考态传播算子方法, 以往的研究表明该方法可以为许多分子开关、分子马达体系中光异构化反应的激发态能量提供可靠的数据. 与CASSCF方法相比, OM2/MRCI方法表现出了一定的优越性. 高精度的从头算CASSCF动力学方法往往局限于较短的模拟时间, 而且由于忽略了不同状态的影响程度、不同的动力学相关性, 从而导致计算处理不平衡[46]. 使用OM2/MRCI轨迹面跳跃法的半经 验动力学计算效率更高, 并且允许计算许多长程动力学且速度更快, 但是作为半经验方法来讲, 它们的固有精度是有限的, 经常需要与高精度从头计 算结果来验证其准确性. 如上所述, 文献[46–50]已证明 OM2/MRCI方法可以正确地描述光诱导过程, OM2/MRCI非绝热动力学可提供静态计算中不明显的机理解释. 与OM2/MRCI方法相比, CASSCF计算激发时, 由于缺乏动力学相关性而容易引起较大的偶极矩变化, 因此CASSCF常常会高估外电荷对激发能的影响. 本文采用多参考OM2/MRCI方法以结合非绝热动力学模拟方法研究了新型吲哚衍生物开关的光致异构化反应机理. 同时计算了光化学反应量子产率、分子体系的态平均寿命、激发态退激途径、荧光辐射谱、红移等现象[9,10,46].
分子的非绝热动力学是从基态E构型(如图1所示)出发, 采用Tully的最少轨迹面跳跃法进行. 模拟计算从 S1 态开始, 模拟总时长设为 500 fs. 其中原子核的演化步长为1 fs, 电子的演化步长则设为 0.001 fs.
为了更好地理解荧光发射光谱, 在图中描述了分子开关的异构化过程期间的时间分辨荧光辐射谱. 在光谱的模拟中, 计算是基于抽样得到的306个动力学轨迹. 为计算出每一时刻的荧光辐射强度, 需要根据从Wigner抽样得到的306个初始轨迹中提取每条轨迹在非绝热动力学计算产生的每时刻的能量及相应的振荡强度、当前态占据数等信息. 然后根据Lan等[51,52] 提出的光谱计算理论基础, 将每一时刻的振荡强度与相应时刻的当前激发态数目的乘积依次累计求和. 为确保计算速度与精度, 计算中我们设置模拟求和步长为10 fs. 下文将展示涉及的光谱计算基本理论公式.
散射截面可以表示为
其中, X和P分别代表分子的原子坐标与动量,
$ Q(X, P, t) $ 为在t时刻的相空间分布函数,$ f(\omega , X, t) $ 为构型X(t)的振荡强度.在任意时刻t, 新型分子开关的含时荧光辐射强度可以表示为
在本文中, 含时分布函数
$ Q(X, P, t) $ 可以用轨迹传播演化来建模. 在任意时刻t, 相关的辐射强度$ {I(\omega , t)}^{{\mathrm{e}}{\mathrm{m}}{\mathrm{i}}{\mathrm{s}}{\mathrm{s}}{\mathrm{i}}{\mathrm{o}}{\mathrm{n}}}{\mathrm{可}}{\mathrm{以}}{\mathrm{表}}{\mathrm{示}}{\mathrm{为}} $ 其中,
$ f\left({\omega }_{i}, t\right) $ 为第i个轨迹在时刻 t时的振荡强度. 需要注意的是, 这里的求和只考虑处于激发态上的轨迹, 因为当分子退激发到基态时是没有辐射的. N1为处于S1激发态的构型数. 为获得光滑的光谱图,$ \delta (\omega -{\omega }_{i}) $ 函数可使用高斯函数进行展宽, 这里归一化的高斯函数可表示为其中,
$ \alpha $ 值设为0.05 eV.时域与频域的辐射强度可以分别表示为
-
在对新型分子开关进行E-Z光致异构化非绝热动力学模拟之前, 首先使用MNDO99软件包中的半经验的OM2/MRCI方法, 对相关基态分子进行结构优化, 优化后的E_S0及Z_S0的基态构型及异构机制如图1所示, 所有原子序号均已在图中标注. 然后通过Winger抽样法获得系列模拟轨迹分子的初始基态分子体系的坐标、速度等信息, 再将分子垂直激发到第一激发态上的FC (Franck-Condon)区域进行500 fs的非绝热动力学模拟. 为深入探究激发态动力学中的分子构型变化, 我们用同样的方法对处于第一激发态的构型进行了优化, 优化后的S1最优构型如图2所示, 结果显示构型发生了明显的改变.
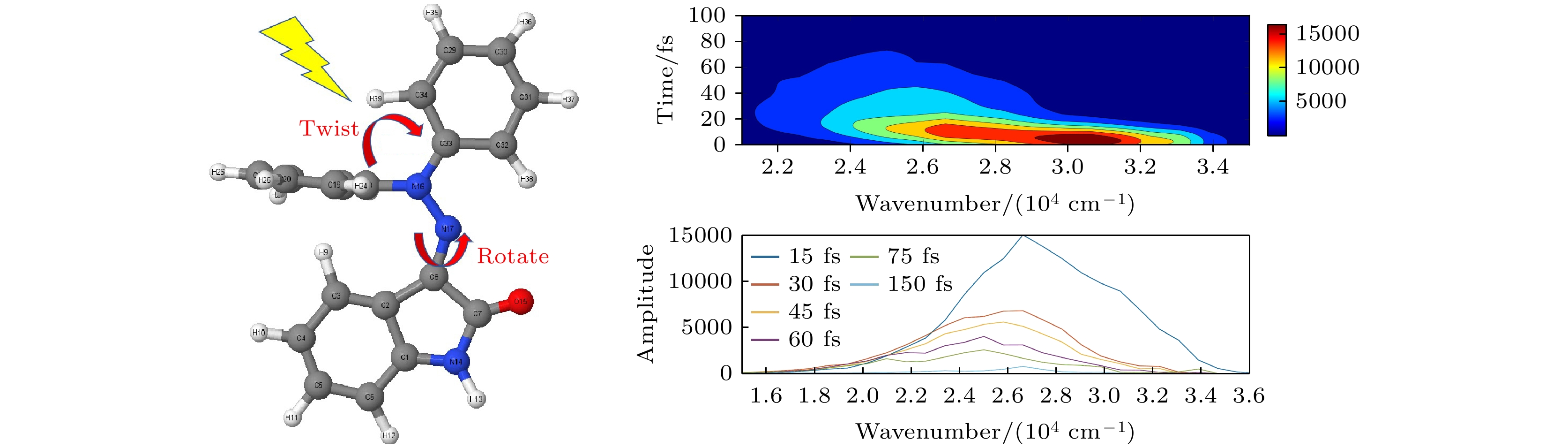
为了准确获得体系在受到光激发后的动力学行为并验证实验上观测到的结果, 对新型分子开关开展了基于Tully的轨迹面跳跃法非绝热动力学模拟研究. 研究结果表明处于激发态的分子体系发生退激时, 主要通过两种异构化方式来生成产物. 一种是传统的围绕中心C=N双键旋转来完成, 另一种是基于对称性双环的扭转来完成.
本文采用OM2/MRCI方法结合Tully的最小面跳跃法, 对从S1激发态FC区域出发的306条轨道进行了500 fs的分子动力学模拟. 在306条轨道中, 275条轨道在500 fs内退激返回到基态, 其中49条轨道经历了异构翻转完成光异构化反应, 这意味着新型靛红双氮二苯腙分子开关光异构化的量子产率约为16.01%. 为了进一步研究靛红双氮二苯腙分子开关退激过程中的电子占据数随时间的变化情况, 我们计算了新型分子开关退激行为中基态和第一激发态电子平均占据数随时间的变化情况.
-
图3中黑线为电子在基态S0的态平均占据数随时间变化曲线, 红线则为电子在第一激发态S1的态平均占据数随时间变化曲线. 从图3可以看到, 在0 fs时, 将基态上的电子垂直激发到第一激发态Franck-Condon区域, 所以在0 fs时刻, 分子在第一激发态S1态上的态平均占据数为1, 而在基态S0的态平均占据数则为0. 在分子发生退激行为的过程中, 我们发现在前28.8 fs电子平均占据数在两个态上几乎没有发生改变, 而在 28.8—312.9 fs 之间, 电子极快地从第一激发态退激到基态, 并且在 107.3 fs 左右时基态和激发态上的电子平均占据数会发生交叉, 基态和激发态电子平均占据数达到均衡, 这也意味着激发态的平均寿命约为 107.3 fs. 在 312.9 fs 以后基本没有基态和激发态电子平均占据数的改变, 意味着激发态的电子已全部退激返回到基态上. 在锥形交叉点, 由于激发态分子发生辐射跃迁, 分子会从两种通道退激从而生成两种产物. 其中, 第一种通道为锥形交叉缝隙左侧, 分子退激发后结构恢复成初始E基态结构; 另一种通道则为锥形交叉缝隙右侧, 此时分子将发生我们熟知的光致异构化行为, 从而生成对应的Z异构体.
为了更详细地理解这两种异构机制, 分别选取了两条不同机制成功发生E-Z异构化的两条典型轨迹进行具体的分析和讨论. 图4(a), (b)代表基于分子特殊的“面对面”转子对称结构, 通过转子部分的扭转来完成异构化的典型轨迹异构机制. 图4(c), (d)则代表通过传统转子相对定子旋转来完成异构化的典型轨迹异构机制. 根据图4(a), (b)可知, 在分子开关扭转异构化过程中, 当分子开关吸收一个光子到达激发态时, 连接定子和转子中心的C8—N17的键长在异构化过程被压缩, 中心转轴C8—N17从0 fs约1.298 Å振荡减小, 在110 fs时其键长达到最小值1.170 Å, 之后在一个稳定值附近来回振荡. 与中心C8—N17键相邻的N16—N17之间原子相互作用被削弱, N16—N17在0 fs从1.328 Å开始有振荡增大的趋势, 在大约在60 fs时其键长最大可以达到约1.483 Å, 去激发后分子开关在一个稳定值附近来回振荡, 与以前研究的分子马达的光驱动异构化机制基本一致. 此外, 我们还研究了与C8—N17相邻的C2—C8的键长变化, 如图4(b), (d)所示, 结果表明在整个异构化过程中C2—C8的键长变化趋势与C8—N17的变化基本保持一致.
图4(c), (d)中当分子开关吸收一个光子到达激发态时, 连接定子和转子中心的C8—N17相互作用被削弱, 中心转轴C8—N17从0 fs约1.338 Å有振荡增大的趋势, 在大约在70 fs时其键长最大可以达到约1.484 Å, 之后在一个稳定值附近来回振荡. 与中心C8—N17键相邻的N16—N17的键长在异构化过程中被压缩, 在0 fs从1.328 Å开始振荡减小, 在大约在45 fs时其键长最大可以达到约1.203 Å, 退激后该键长在一个稳定值附近来回振荡. 同时, C8—N17与C2—C8的键长变化趋势基本保持一致.
其中, 最引人注目的是二面角的时间变化. 同理, 我们对选取的两条典型轨迹进行详细的机制分析. 图5(a), (b)代表基于分子特殊的“面对面”转子对称结构, 通过转子部分的扭转来完成异构化的典型轨迹异构机制. 图5(c), (d)则代表通过传统转子相对定子旋转来完成异构化的典型轨迹异构机制. 从二面角C8—N17—N16—C33与C8—N17—N16—C18 的图像变化趋势(图5(a), (b))可以得出二者的变化规律大致相同, 在0—500 fs, 两个二面角都向着负方向发生极快的旋转, 其角度先从反方向上减小, C8—N17—N16—C33在165.4 fs时达到反方向上角度的最小值, 减小为0°, 而C8—N17—N16—C18随后在反方向上一直增大, 在218.8 fs时达到反方向上角度的最小值, 减小为0°, 二者在到达0°以后在反方向上一直增大. 值得注意的是, 二者在140—270 fs急剧下降, 进行了快速扭转. 二面角C2—C8—N17—N16, C7—C8—N17—N1的变化规律与C8—N17—N16—C33, C8—N17—N16—C18相反, 很明显该二面角反转了180°. 研究表明在整个异构化过程中两个二面角的旋转起到了非常关键的作用. 计算结果表明在分子开关的异构化过程中, 上半部分两个转子部分通过旋转都参与了异构化反应, 这也是第二代新型分子开关的第一种异构化方式-扭转.
图5(c), (d)描述的旋转方式是以C=N为中心含O原子的下半部分(单个转子)旋转360°, 通过图5(d)的图像可知, 二面角 C8—N17—N16—C18发生了明显的旋转, 在前135 fs, 二面角向着正方向发生了极快的旋转, 随后在135—235 fs之内, 其角度来回振荡并在 235 fs时增大到最大值108.0°, 随后角度则减小, 逐渐趋于稳定. 如图5(c)所示, 二面角 C2—C8—N17—N16的角度从180°减小至0°; C7—C8—N17—N16的角度从0°减小至–180°; 而C8—N17—N16—C33 的变化不太明显, 通过以上4个二面角的数据分析, 可以清楚观察到该分子开关成功旋转180°, 异构化成功. 计算结果表明在分子开关的异构化过程中, 下半部分的转子通过旋转都参与了异构化反应, 这也是第二代新型分子开关的第二种异构化方式.
-
基于振荡强度计算的荧光发射的二维轮廓如图6所示. 图6(a)中x和y轴分别是波数和时间. 如图6(a)所示, 深红色的中心波数代表荧光光谱的峰值强度, 计算的荧光发射光谱在75 fs左右消失, 反映了荧光辐射的极快猝灭发生在75 fs之前. 此外, 图6(b)中x和y轴分别是波数和振幅, 图中不同颜色表示15—150 fs时间范围内的波长分辨荧光辐射谱随时间变化的曲线, 振幅代表不同频率振荡强度. 从图中可以看出, 其振幅振荡强度随时间的增加而逐渐减小, 并伴随着辐射光子波数的红移. 在15 fs处荧光光谱的波数约为2.6 × 104 cm–1, 而75 fs处的约为1.6× 104 cm–1, 与15 fs处的荧光光谱相比, 其强度要弱得多. 结合图6(a), (b), 可以清楚地看到荧光红移现象主要发生在2.1×104—3.4×104 cm–1波段处. 因此, 荧光辐射光谱红移和猝灭发生在该分子开关系统中. 由于荧光辐射的寿命约为75 fs, 比 S1激发态的寿命(约107 fs)短, 因此理论预测在该新型分子开关的光异构化过程中可能存在“暗态”现象. 需要强调的是, 该“暗态”可能并不是新的电子传输态, 而是处于S1态上的一段特殊的区域, 该范围内分子的S1→S0振荡因子相对比较低[33].
-
本文采用基于OM2/MRCI水平的面跳跃非绝热分子动力学模拟, 研究了新型分子开关的非绝热光致异构化动力学. 根据计算结果, 得到该分子开关的E-Z异构化量子产率为16.01%, 光异构化过程的平均S1激发态寿命为107.3 fs, 表明新型分子开关在异构化过程中的光致异构化动力学都处于超快时间尺度. 模拟表明, 在S0→S1激发后, 分子会通过两种不同的机制发生异构化反应, 一种是常见的围绕中心C=N双键的旋转发生的异构化反应, 另一种是基于分子开关上半部分特殊的对称性结构, 直接发生上半部分的扭转异构化来完成整体的异构化反应. 此外, 通过计算随时间演化的荧光辐射谱, 预测了该分子开关在退激过发程中会发生极快的荧光猝灭现象, 时间大约发生在75 fs, 因此, 计算预测该分子开关在发生异构化反应时会存在短暂的“暗态”现象. 同时, 计算中观察到了荧光辐射伴随着波的“红移”现象. 这可能与分子开关的异构化量子产率有间接性的关联, 通过修饰分子开关的构型削弱“暗态”存在的时长或者减弱红移现象可能有利于提高其光致异构化量子产率. 该工作为设计一系列新型高效分子开关奠定了理论基础.
-
本篇论文的关联数据可在科学数据银行
https:// doi.org/10.57760/sciencedb.11735 中访问获取.
靛红双氮二苯腙分子开关的光致异构化机理
Photoinduced isomerization mechanism of isatin N2-diphenylhydrazones molecular switch
-
摘要: 腙类分子开关在超分子化学领域有非常重要的应用价值, 基于靛红发色团发展出的新型腙类分子开关已被合成. 因其具有可见光激发下的顺反异构化特征、衍生物的易合成性及对外界刺激的敏感性, 其在生物化学领域具有重要的应用价值. 然而, 该新型分子开关的光致异构化机制尚不明确, 异构过程是否存在新奇的现象也不得而知. 本文采用基于半经验OM2/MRCI的轨迹面跳跃动力学方法, 系统地研究了一种名为靛红双氮二苯腙分子开关E-Z异构化过程的光致异构化机理. 研究发现该E构型分子开关的S1激发态平均寿命约为 107 fs, 该分子开关的E-Z异构化量子产率为16.01%. 通过对该分子开关光致异构化过程的计算, 明确了两种不同分子开关异构化机制; 除围绕C=N键旋转的传统分子开关异构机制之外, 阐明了新的异构机制——分子开关转子部分面对面的扭转; 通过时间分辨的荧光辐射谱的计算, 预测了异构化过程中存在极快的荧光猝灭现象, 并且伴随着荧光红移的发生; 通过对计算得到的荧光光谱与激发态平均寿命的分析, 提出了“暗态”的存在, 并对“暗态”存在的原因给出了可能的解释. 研究结果可为新型分子开关的设计及应用提供理论指导作用.Abstract: Hydrazone molecular switches have significant application value in supramolecular chemistry. A new type of hydrazone molecular switch, named isatin N2-diphenylhydrazone, has been synthesized. Owing to its cis-trans isomerization characteristics under visible light excitation, ease of synthesizing of derivatives, and sensitivity to external stimuli, it has important application value in the field of biochemistry. Because of its forward and backward visible light excitation characteristics, it is considered a class of compound that is very suitable for molecular switches, and it has a wide application value in fields such as biotechnology. In addition, the derivatives compound exhibits strong interactions with negative ions, which enhances its function as a molecular switch, making it a four-state molecular switch that can be achieved by a single molecule. However, the photo-induced isomerization mechanism of these new molecular switches is not yet clear, and whether there are novel phenomena in the isomerization process is also unknown. In this work, a semi empirical OM2/MRCI based trajectory surface hopping dynamics method is adopted to systematically study a photo induced isomerization mechanism based on the E-Z isomerization process of the isatin N2-diphenylhydrazones molecular switch. Optimization configuration and the average lifetime of the first excited S1 state are obtained by using the semi-empirical OM2/MRCI method of molecular switch. It is found that the average lifetime of the S1 excited state of the E-configuration molecular switch is about 107 fs, and the quantum yield of E-Z isomerization of the molecular switch is 16.01%. By calculating the photo induced isomerization process of the molecular switch, two different isomerization mechanisms of the molecular switch are identified. In addition to the traditional molecular switch isomerization mechanism revolving around the C=N bond, a new isomerization mechanism, i.e. the face-to-face twisting of the molecular switch rotor part is elucidated. By calculating the time-resolved fluorescence radiation spectrum, it is predicted that there may be a very fast fluorescence quenching phenomenon occurring in about 75 fs in the isomerization process, slightly faster than the S1 average decay events (107 fs). The information about wavelength-resolved attenuation at different times is also calculated, which reflects the ultrafast fluorescence quenching process accompanied by fluorescence red shift, ranging from 2.1 × 104 cm–1 to 3.4 × 104 cm–1. By comparing the calculated fluorescence spectra with the average lifetime of excited states, the existence of “dark states” is proposed, and possible explanations for the existence of “dark states” are provided, and those “dark states” may be related to lower quantum yields. The research results can provide theoretical guidance for the design and application of new molecular switches. The ease of synthesis and sensitivity to external stimuli of its derivatives make those compounds extremely valuable in molecular switching and light measurement applications.
-
Key words:
- molecular switch /
- photoinduced isomerization /
- excited state /
- non adiabatic dynamics .
-

-
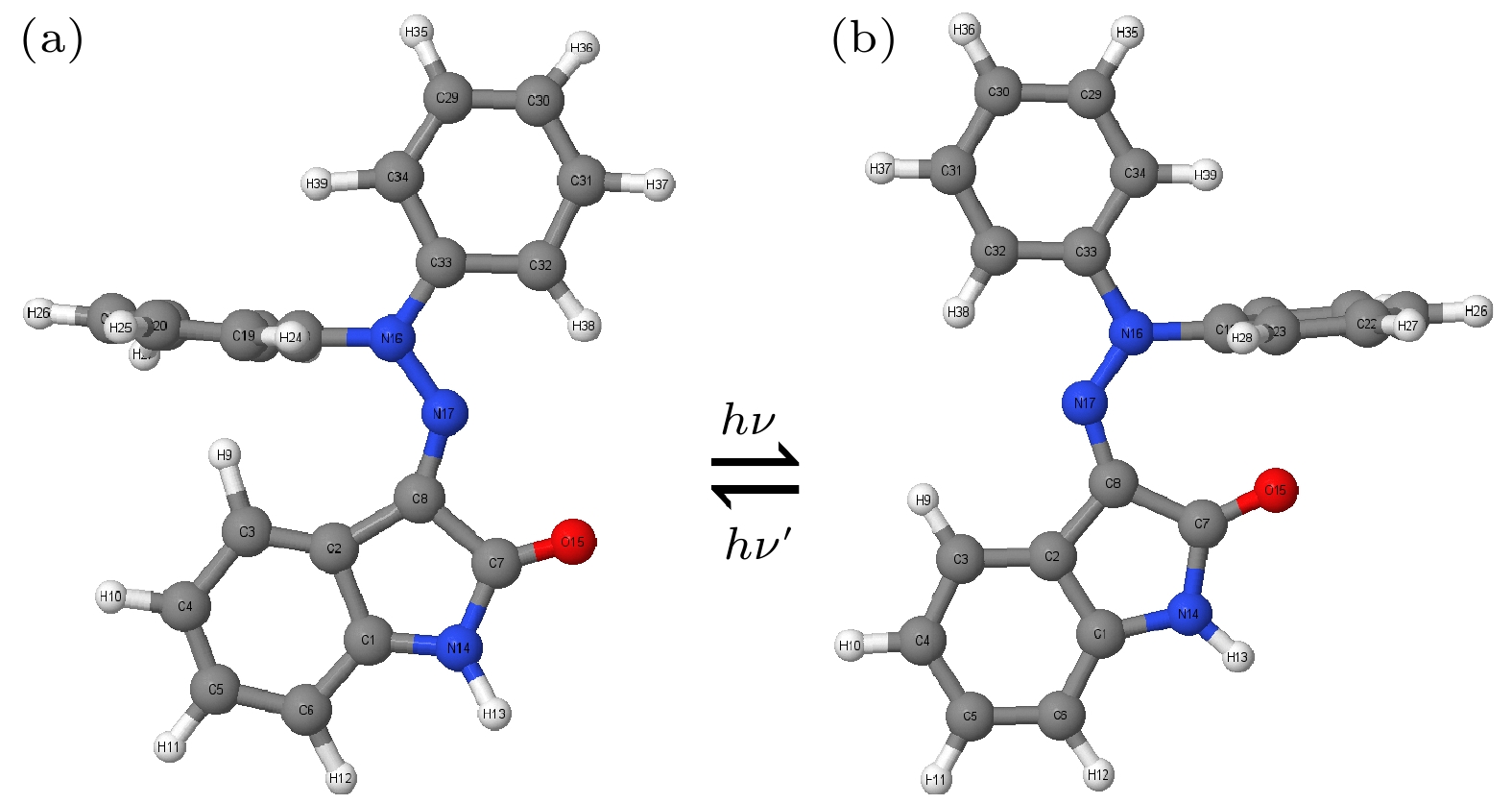
图 1 新型分子开关E-Z异构机制图, 最优基态反应物 (a) E及产物(b) Z的S0最优构型图, 所有相关原子序号均已标注
Figure 1. E-Z isomerization mechanism diagram of a novel molecular switch, the optimal configuration diagram of S0 for the optimal ground state reactant (a) E and product (b) Z, with all relevant atomic numbers marked.
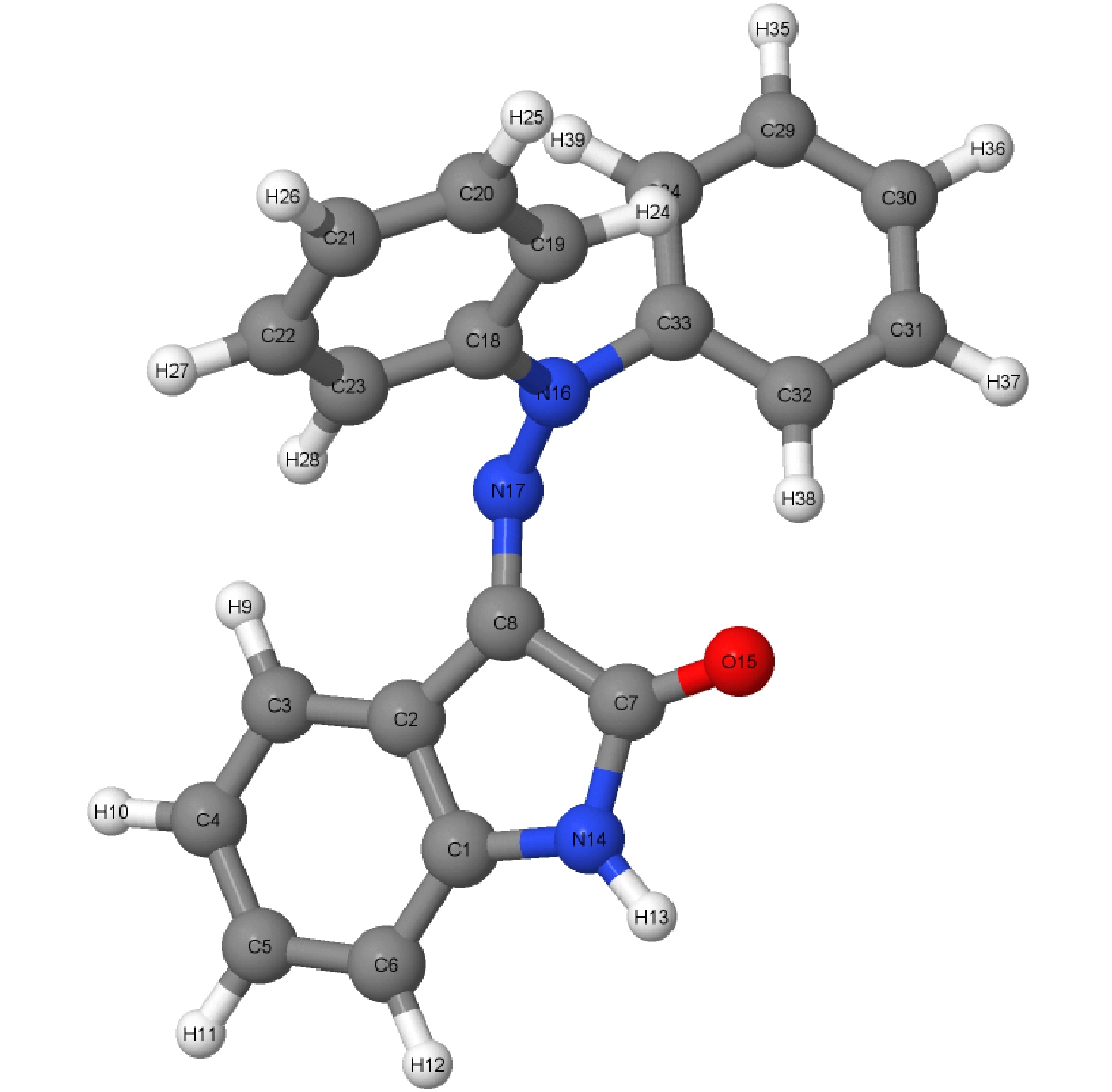
图 2 新型分子开关半经验 OM2/MRCI方法下优化的第一激发态S1最优构型
Figure 2. Optimization of the optimal configuration of the first excited state S1 under the semi-empirical OM2/MRCI method of the novel molecular switch.
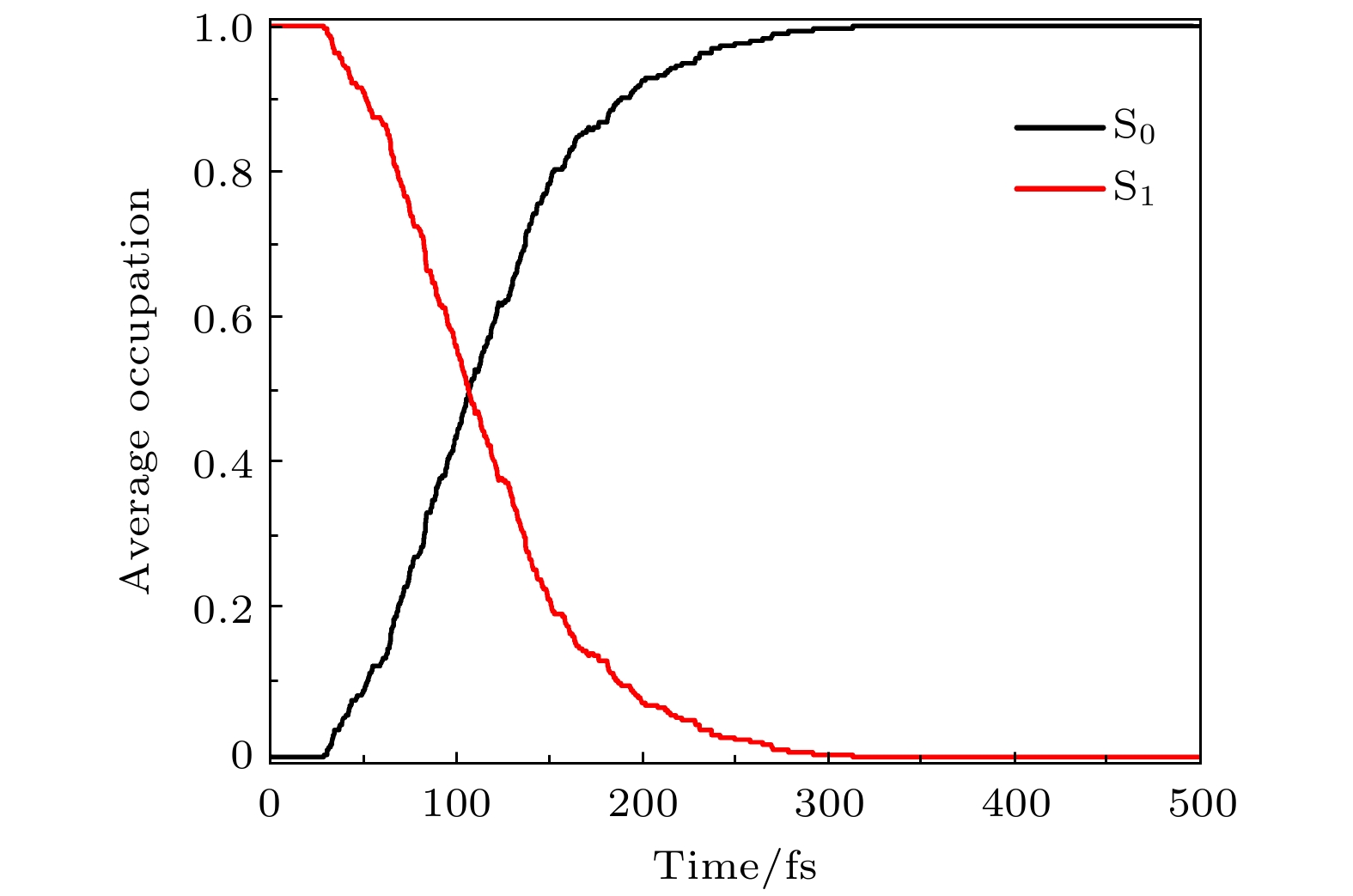
图 3 电子在基态S0和第一激发态S1的态平均占据数随时间变化情况
Figure 3. The average occupancy of electrons in the ground state S0 and the first excited state S1 over time.
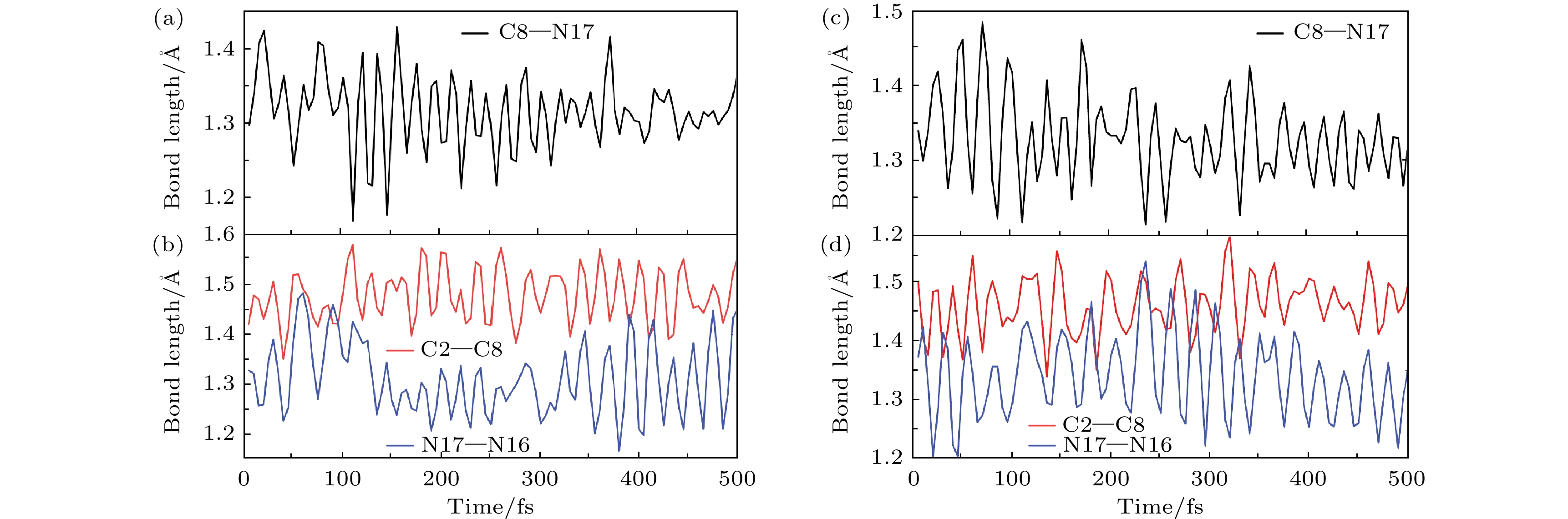
图 4 新型分子开关异构化过程中的键长的变化趋势图 (a), (b)扭转过程中C8—N17 与C2—C8, N17—N16的变化趋势; (c), (d)旋转过程中C8—N17 与C2—C8, N17—N16变化趋势
Figure 4. Trend chart of bond length changes during the isomerization process of the novel molecular switch: (a), (b) The changing trends of C8—N17 and C2—C8, N17—N16 during the twisting process; (c), (d) changes in C8—N17 and C2—C8, N17—N16 during rotation.
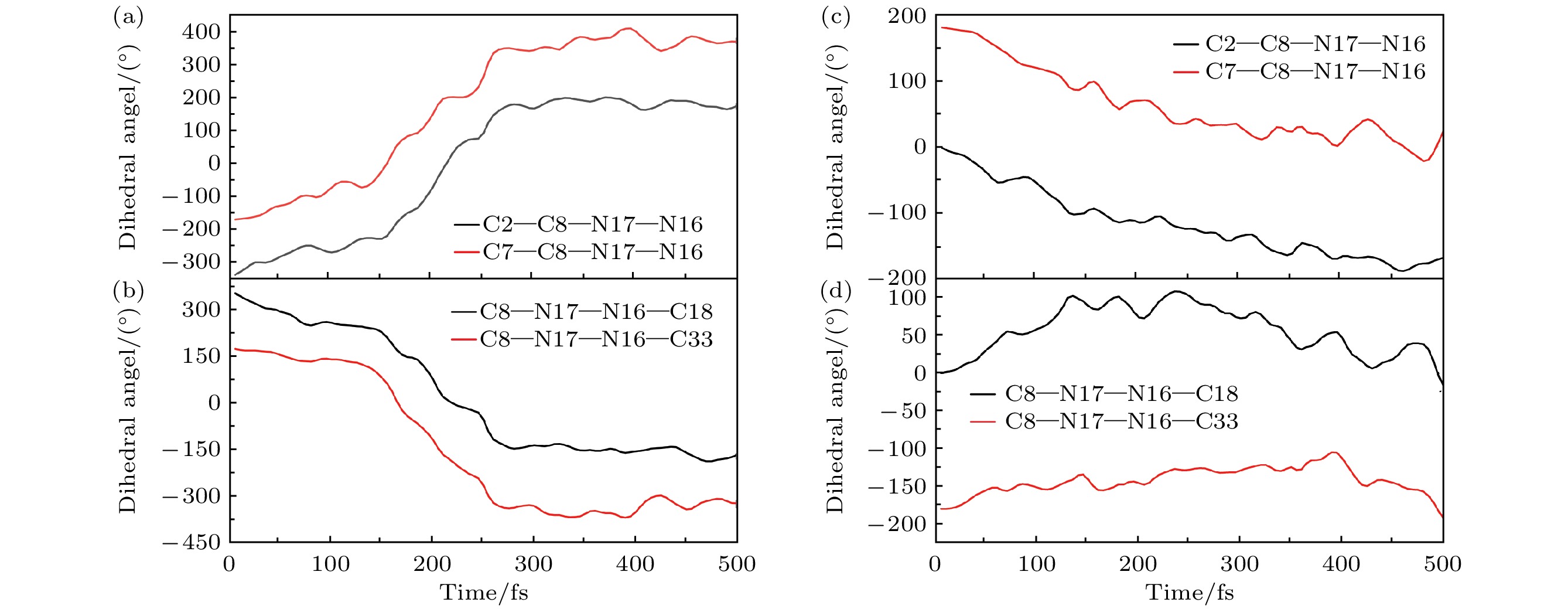
图 5 新型分子开关异构化过程中的二面角的变化趋势 (a), (b)扭转过程中C2—C8—N17—N16, C7—C8—N17—N16与C8—N17—N16—C33, C8—N17—N16—C18的变化趋势; (c), (d)旋转过程中C2—C8—N17—N16, C7—C8—N17—N16与C8—N17—N16—C33, C8—N17—N16—C18的变化趋势
Figure 5. The changing trend of dihedral angle during the isomerization process of the novel molecular switch: (a), (b) The changing trends of C2—C8—N17—N16, C7—C8—N17—N16, C8—N17—N16—C33, and C8—N17—N16—C18 during the twisting process; (c), (d) the changing trends of C2—C8—N17—N16, C7—C8—N17—N16, C8—N17—N16—C33, and C8—N17—N16—C18 during the rotation process.
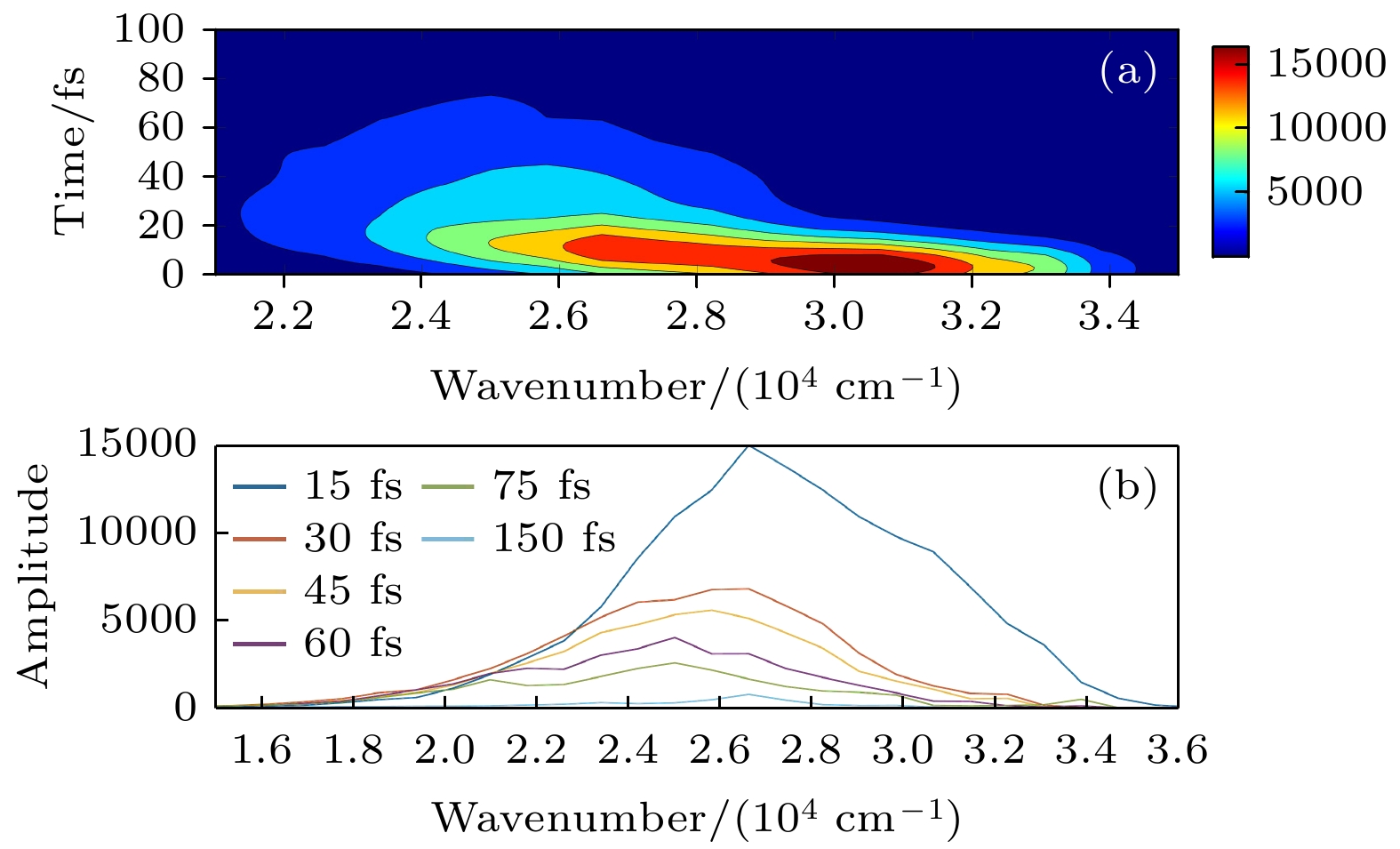
图 6 激发态动力学的含时荧光辐射光谱 (a)随时间变化荧光辐射光谱; (b)不同时间下随波长变化的衰减信息
Figure 6. Time-dependent fluorescence emission spectra of excited state dynamics: (a) The time-resolved deconvoluted emission spectrum; (b) the wavelength-resolved decay information in different time.
-
[1] Li R J, Mou B Z, Yamada M, Li W, Nakashima T, Kawai T 2024 Molecules 29 25 doi: 10.1097/01.bor.0000240371.85454.29 [2] Jago D, Gaschk E E, Koutsantonis G A 2023 Aust. J. Chem. 76 635 doi: 10.1071/CH23115 [3] Yu Z, Hecht S 2016 Chem. Commun. 52 6639 doi: 10.1039/C6CC01423B [4] Feringa B L, Van Delden R A, Koumura N, Geertsema E M 2000 Chem. Rev. 100 1789 doi: 10.1021/cr9900228 [5] Rice A M, Martin C R, Galitskiy V A, Berseneva A A, Leith G A, Shustova N B 2020 Chem. Rev. 120 8790 doi: 10.1021/acs.chemrev.9b00350 [6] Zhang X Y, Hou L L, Samorì P 2016 Nat. Commun. 7 11118 doi: 10.1038/ncomms11118 [7] Alenazi M H, Mubarak A T, Abboud M 2024 Nanotechnol. Rev. 13 20240032 doi: 10.1515/ntrev-2024-0032 [8] Goulet-Hanssens A, Eisenreich F, Hecht S 2020 Adv. Mater. 32 1905966 doi: 10.1002/adma.201905966 [9] Pios S V, Gelin M F, Ullah A, Dral P O, Chen L 2024 J. Phys. Chem. Lett. 15 2325 doi: 10.1021/acs.jpclett.4c00107 [10] Conti I, Cerullo G, Nenov A, Garavelli M 2020 J. Am. Chem. Soc. 142 16117 doi: 10.1021/jacs.0c04952 [11] Towns A 2021 Phys. Sci. Rev. 6 477 doi: 10.1515/psr-2020-0191 [12] Cheng H B, Zhang S, Bai E, Cao X, Wang J, Qi J, Liu J, Zhao J, Zhang L, Yoon J 2022 Adv. Mater. 34 2108289 doi: 10.1002/adma.202108289 [13] Bertarelli C, Bianco A, Castagna R, Pariani G 2011 J. Photoch. Photobio. C 12 106 doi: 10.1016/j.jphotochemrev.2011.05.003 [14] Bléger D, Hecht S 2015 Angew. Chem. Int. Ed. 54 11338 doi: 10.1002/anie.201500628 [15] Shao B, Aprahamian I 2020 Chem 6 2162 doi: 10.1016/j.chempr.2020.08.007 [16] Van Dijken D J, KovaříčEk P, Ihrig S P, Hecht S 2015 J. Am. Chem. Soc. 137 14982 doi: 10.1021/jacs.5b09519 [17] Schnetz M, Meier J K, Rehwald C, Mertens C, Urbschat A, Tomat E, Akam E A, Baer P, Roos F C, Brüne B 2020 Cancers 12 530 doi: 10.3390/cancers12030530 [18] Ferreira I P, Piló E D, Recio-Despaigne A A, Da Silva J G, Ramos J P, Marques L B, Prazeres P H, Takahashi J A, Souza-Fagundes E M, Rocha W 2016 Bioorg. Med. Chem. 24 2988 doi: 10.1016/j.bmc.2016.05.007 [19] Vantomme G, Lehn J M 2014 Chem. Eur. J. 20 16188 doi: 10.1002/chem.201404561 [20] Vantomme G, Lehn J M 2013 Angew. Chem. Int. Edit. 52 3940 doi: 10.1002/anie.201210334 [21] Vantomme G, Jiang S M, Lehn J M 2015 J. Am. Chem. Soc. 137 3138 doi: 10.1021/jacs.5b00419 [22] Su X, Aprahamian I 2014 Chem. Soc. Rev. 43 1963 doi: 10.1039/c3cs60385g [23] Chaur M N, Collado D, Lehn J M 2011 Chem. Eur. J. 17 248 doi: 10.1002/chem.201002308 [24] Vantomme G, Jiang S, Lehn J M 2014 J. Am. Chem. Soc. 136 9509 doi: 10.1021/ja504813r [25] 刘同力, 黄从树, 王晶晶, 梁宇, 谢志鹏, 庄海燕, 李九龙, 朱绪飞 2024 精细化工 https://doi.org/10.13550/j.jxhg.20230989 Liu T L, Huang C S, Wang J J, Liang Y, Xie Z P, Zhuang H Y, Li J L, Zhu X F 2024 Fine. Chem https://doi.org/ 10.13550/ j.jxhg.20230989 [26] Siewertsen R, Neumann H, Buchheim-Stehn B, Herges R, Näther C, Renth F, Temps F 2009 J. Am. Chem. Soc. 131 15594 doi: 10.1021/ja906547d [27] Beharry A A, Sadovski O, Woolley G A 2011 J. Am. Chem. Soc. 133 19684 doi: 10.1021/ja209239m [28] Poloni C, Szymanski W, Hou L L, Browne W R, Feringa B L 2014 Chem. Eur. J. 20 946 doi: 10.1002/chem.201304129 [29] Zhang Z W, Yang W X, Zhang J J 2023 Chem. Ind. Eng. Prog 42 4058 [张志伟, 杨伟鑫, 张隽佶 2023 化工进展 42 4058] doi: 10.16085/j.issn.1000-6613.2023-0894 Zhang Z W, Yang W X, Zhang J J 2023 Chem. Ind. Eng. Prog 42 4058 doi: 10.16085/j.issn.1000-6613.2023-0894 [30] Ye H 2020 M. S. Thesie (Wuhan: Huazhong University of Science and Technology) [叶欢 2020 硕士学位论文(武汉: 华中科技大学)] Ye H 2020 M. S. Thesie (Wuhan: Huazhong University of Science and Technology) [31] Szymanski W, Beierle J M, Kistemaker H A, Velema W A, Feringa B L 2013 Chem. Rev. 113 6114 doi: 10.1021/cr300179f [32] Cigán M, Gáplovsky M, Jakusová K, Donovalová J, Horváth M, Filo J, Gáplovsky A 2015 RSC Adv. 5 62449 doi: 10.1039/C5RA06625E [33] Cigáň M, Jakusová K, Gáplovský M, Filo J, Donovalová J, Gáplovský A 2015 Photochem. Photobiol. Sci. 14 2064 doi: 10.1039/c5pp00275c [34] Tisovský P, Donovalová J, Kožíšek J, Horváth M, Gáplovský A 2022 J. Photochem. Photobiol. A Chem. 427 113827 doi: 10.1016/j.jphotochem.2022.113827 [35] Seleem H S 2011 Chem. Cent. J. 5 8 doi: 10.1186/1752-153X-5-8 [36] Šandrik R, Tisovský P, Csicsai K, Donovalová J, Gáplovský M, Sokolík R, Filo J, Gáplovský A 2019 Molecules 24 2668 doi: 10.3390/molecules24142668 [37] Liu H H, Chen Y 2009 J. Phys. Chem. A 113 5550 doi: 10.1021/jp810919j [38] Tochitsky I, Polosukhina A, Degtyar V E, Gallerani N, Smith C M, Friedman A, Van Gelder R N, Trauner D, Kaufer D, Kramer R H 2014 Neuron 81 800 doi: 10.1016/j.neuron.2014.01.003 [39] Van Herpt J T, Areephong J, Stuart M C, Browne W R, Feringa B L 2014 Chem. Eur. J. 20 1737 doi: 10.1002/chem.201302902 [40] Nakagawa T, Ubukata T, Yokoyama Y 2018 J. Photochem. Photobiol. C-Photochem. Rev. 34 152 doi: 10.1016/j.jphotochemrev.2017.12.004 [41] Kistemaker J C M, Stacko P, Roke D, Wolters A T, Heideman G H, Chang M C, Van Der Meulen P, Visser J, Otten E, Feringa B L 2017 J. Am. Chem. Soc. 139 9650 doi: 10.1021/jacs.7b04412 [42] Kistemaker J C M, Stacko P, Visser J, Feringa B L 2015 Nature Chemistry 7 890 doi: 10.1038/nchem.2362 [43] Thiel W 1981 J. Am. Chem. Soc. 103 1413 doi: 10.1021/ja00396a021 [44] Wang J, Durbeej B 2018 ChemistryOpen 7 583 doi: 10.1002/open.201800089 [45] Ma J Z, Yang S J, Zhao D, Jiang C W, Lan Z G, Li F L 2022 Int. J. Mol. Sci. 23 3908 doi: 10.3390/ijms23073908 [46] Pang X J, He H Y, Zhao K Y, Zhang N B, Zhong Q J 2023 Chem. Phys. Lett. 819 140439 doi: 10.1016/j.cplett.2023.140439 [47] Zhuang X H, Wang J, Lan Z G 2013 J. Phys. Chem. A 117 4785 doi: 10.1021/jp402180p [48] Pang X J, Cui X Y, Hu D P, Jiang C W, Zhao D, Lan Z G, Li F L 2017 J. Phys. Chem. A 121 1240 doi: 10.1021/acs.jpca.6b12253 [49] Weber W, Thiel W 2000 Theor. Chem. Acc. 103 495 doi: 10.1007/s002149900083 [50] Otte N, Scholten M, Thiel W 2007 J. Phys. Chem. A 111 5751 doi: 10.1021/jp0700130 [51] Lan Z G, Lu Y, Weingart O, Thiel W 2012 J. Phys. Chem. A 116 1510 doi: 10.1021/jp2117888 [52] Jin H, Liang M, Arzhantsev S, Li X, Maroncelli M 2010 J. Phys. Chem. B 114 7565 doi: 10.1021/jp100908a -
计量
- 文章访问数: 895
- HTML全文浏览数: 895
- PDF下载数: 4
- 施引文献: 0