
 首页
首页 登录
登录 注册
注册



 下载:
下载:
-
界面动态相互作用是生物物理及相关研究领域的一个核心关键问题. 一个典型例子是生物活性分子(如蛋白质、多肽、聚合物分子和纳米颗粒)与细胞膜之间的相互作用[1–6]. 作为将细胞内部与外部环境隔离开来的物理屏障, 细胞膜参与了许多基本的生物学过程, 包括内吞作用、外排作用、离子运输、信号转导、细胞迁移以及病毒入侵和自体防御肽杀菌等[7–11]. 此外, 目前超过60%的药物通过靶向细胞膜上特定蛋白质或脂质结构发挥功效. 同时, 这种生物-纳米界面作用与生物毒性现象以及纳米材料实现生物功能密切相关[12,13]. 因此, 实时且无创地研究生物活性分子与细胞膜之间的相互作用对于理解和调控这些生物学过程中所涉及的分子机制, 并指导新型药物或药物载体设计具有重要意义[14–17].
为了实现这一目的, 研究者们采用了多种实验技术. 其中最常用的方法之一是圆二色谱光谱技术, 其通过测量光活性物质对左旋和右旋圆偏振光的吸收差异, 快速获取生物大分子(如蛋白质和多肽)的二级结构信息[18–20]. 因此, 圆二色谱方法通常应用于监测生物大分子与细胞膜相互作用过程中的二级结构变化. 其他一些分子光谱方法, 如紫外-可见吸收光谱、红外光谱或拉曼光谱, 也是提取具有高时间或结构分辨率中间态信息的有力工具. 然而, 这些方法可能无法准确描述膜结构(或脂质排列状态)的微观或宏观特征. 除了上述光谱技术外, 光学显微镜成像是生物研究中另一类传统方法. 然而即使在开发了新型的共聚焦、高分辨率或超分辨率技术情况下, 空间和/或时间分辨率有限等缺点仍然存在, 这也使得很难提供细胞膜(厚度仅为4—5 nm)界面上的分子水平信息[21]. 近年来, 荧光共振能量转移(FRET)、表面引起的荧光衰逝(SIFA)和单分子追踪等基于荧光定位和成像的技术在得到了快速发展[22–25]. 然而, 这些测试需要对待测样品进行荧光探针标记, 可能会干扰待测分子的行为, 并且测试过程对生物体系带来的光毒性也是不容忽视的问题. 相比之下, 电子显微镜技术如扫描电子显微镜(SEM)或透射电子显微镜(TEM), 特别是冷冻TEM, 则可以提供更多关于膜和蛋白质结构的信息[26–30]. 例如, 在分析囊泡膜复合结构方面已经取得成功[31,32]. 但它们缺乏实时非侵入性检测能力, 并且样品制备过程中的复杂操作可能会影响观察结果. 另一方面, 跨膜电测量为监测细胞膜动态过程提供了无标签、实时和高灵敏度的方法. 例如, 电化学工作站可以直接用于磷脂膜的稳态电流测量, 而膜片钳法则常被用于研究细胞膜离子通道[33,34]. 但这些方法需要在系统中施加偏电场, 可能会干扰生物过程特别是一些带电分子的生物功能, 如抗菌肽和纳米颗粒. 在过去几十年中, 二次谐波产生(SHG)与和频产生(SFG)等非线性光学技术得到了广泛发展. 这些方法逐步提高了空间(例如在分子水平上)和时间分辨率(例如, 飞秒级别的SFG时间分辨率小于1 s), 并被应用于表征界面结构和相互作用研究[35–37]. 由于这些方法源自诱导极化而非实际吸收, 并采用非共振光学相互作用, 因此有望成为无创、低光毒性且适用于生物界面原位成像的备选方案. 总之, 所有实验技术都具有各自的优点和局限性. 对于实时监测生物界面动态过程来说, 开发原位、无创、无标记、灵敏且易操作的技术至关重要, 但也是目前大多数现有技术所面临的挑战.
针对这一问题, 我们与合作者开发了一种新型的光电压瞬态技术, 用于实时监测人工细胞膜界面动态过程[38], 并广泛应用于不同材料与膜相互作用过程的研究, 包括表面活性剂[39]、聚合物[40]、多肽[41–44]和纳米颗粒[45]等. 光电压瞬态技术是利用半导体材料的光电响应来获取光致电子, 并通过跟踪这些光致电子在细胞膜内部的充放电过程及其随时间变化情况来获取分子与膜相互作用过程中细胞膜结构和性质发生变化的动态信息. 该技术具有百毫秒级别的时间分辨率, 在宏观尺度上能够检测到整个膜的全貌信息, 并具备原位、无创、灵敏且无需标记等特点; 并且, 光电压瞬态设备搭建成本低廉(5—10万元人民币), 易操作, 也无需复杂的数据处理过程, 因此, 该技术为实现细胞膜等重要生物界面的实时、原位监测提供了备选途径. 本综述将系统阐述光电压瞬态技术的工作原理、设备搭建以及数据处理方法, 并以最具代表性的细胞膜模型-磷脂双层膜为例, 概述该技术在探索磷脂膜水合特性及其与活性分子(如表面活性剂、聚合物、多肽和纳米颗粒)相互作用机制方面取得的最新进展. 最后就该技术优缺点进行讨论, 并展望未来发展前景.
-
半导体材料(例如硅片)的光电响应在光激活时提供了一种可控和非破坏性的电子流, 这使得对膜复杂结构变化的非接触实时监测成为可能. 将细胞膜铺展在硅片表面, 并置于含有活性分子的低离子水溶液中作为“光电极”(即“工作电极”). 通过记录光致电子在细胞膜内积累后衰减过程(即充放电过程), 可以直接获取膜瞬时结构和性质信息, 这一过程可通过工作电极与参比电极之间的电压差进行精确记录. 实时记录随时间变化的“充放电曲线”, 能够揭示膜在周围分子/溶液作用下发生的变化. 例如, 在硅片表面沉积连续磷脂双分子层模拟细胞膜, 并利用方波调制光源进行照射; 由于磷脂膜是一种电介质, 其与溶液环境界面处形成的双电层会产生相应的充放电过程, 该过程可以被记录为输出电压曲线; 如果出现跨膜穿孔等结构扰动, 则会显着影响这种充放电曲线, 并且相关信号将被及时捕捉到. 通过分析随时间变化的充放电曲线, 可以原位、实时、准确地监测分子诱导引起磷脂膜结构变化, 并进一步了解分子-膜相互作用动态过程.
在该技术中存在几个先决条件: 首先, 电路中的电流值应接近于零(例如,
$\ll $ 1 nA). 只有在这种情况下, 输出电压的变化才能准确反映膜中剩余电荷的变化, 并进一步揭示膜结构和性质的变化. 其次, 测量过程不应对分子与膜之间的相互作用产生干扰. 此外, 需要较大的照明面积以确保宏观尺度上检测到整个膜信息, 例如分子吸附和逐渐聚集于膜表面以及跨膜孔形成等过程. -
光电压测试系统包含以下模块(见图1): 置于电磁屏蔽箱内的反应仓(包括工作电极和参比电极), 信号放大模块, 信号采集模块, 可控光源以及用于操控和收集处理信号的计算机. 反应仓被安置在避光且具备电磁屏蔽功能的箱体中. 一台功率为500 W、波长范围为300—1500 nm的氙灯提供宽谱连续光源, 确保稳定照明; 单色仪被安装在光路上, 用于研究不同波长下光电极的响应特性. 通过计算机控制氙灯产生方波调制光来激发并照亮反应仓中的光电极以实现其光电响应. 采用带有石英窗的聚四氟乙烯(PTFE)室作为反应仓. 该室分为两个相互隔离部分, 用于控制溶液混合. 将反应仓安装在二维滑动台上, 以调节光电极上的照射面积. 为了方便操作和提高重复性, 设计了一套功能性夹具来固定整个反应仓, 并在整个测试过程中保持系统的稳定性. 此外, 还设计了专用的电极适配板, 通过探头与光电极良好接触, 并将信号传输到高频连接器. 输出电压信号经放大模块放大后, 由信号采集模块转换为数字信号, 并由计算机进行收集. 计算机对数据进行处理以分析光电极界面上发生的动态过程.
该系统的关键部件是工作电极(又称光电极, 即待测体系), 它与参比电极(标准Ag/AgCl电极)一起浸入反应仓的溶液中. 工作电极由导电掺杂硅片以及硅片表面覆盖的磷脂膜组成, 硅片在水中的无膜区域经过绝缘化处理. 在光照下, 硅片发生光电效应, 使得光致电荷在电介质膜内积累和衰减.
-
为了制备工作电极, 采用硅片支撑的磷脂酰胆碱(1, 2-dioleoyl-sn-glycero-3-phosphocholine, DOPC, 购于Avanti Polar Lipids)单层(supported lipid bilayer, SLB)或多层(multilayered lipid bilayer, MLB)磷脂膜, 它们皆是最为常用的细胞膜模型. 首先切割硅片(N型重掺, 面积为3.5 cm×1 cm)并进行彻底清洁, 同时保持表面的亲水性. 接下来, 使用传统的囊泡融合法制备SLB, 将DOPC磷脂溶解在氯仿中(浓度为10.0 mg/mL), 然后在N2流中完全干燥. 随后, 在纯水中重新水合(浓度为3.5 mg/mL), 经过超声处理1 h, 并通过具有100 nm孔径的聚碳酸酯膜(avanti polar lipids)挤压21次, 即可得到分散性良好的单层小囊泡(small unilamellar vesicles, SUVs). 根据动态光散射测量结果显示SUVs尺寸约为(110 ± 15) nm. 将洁净的硅片表面一端(覆盖面积为2 cm×1 cm)浸泡于SUVs分散液中并孵育3 h, 然后小心地冲洗掉多余物质. 由于硅表面具有亲水性, 在这个过程中能够形成高质量的SLB结构. 样品在使用前最多保存10 min且相对湿度不低于90%, 以保持双层结构稳定. 在对照实验中(包含摩尔分数为0.5%的荧光标记脂质), 通过原子力显微镜技术(AFM)和共聚焦荧光显微镜光漂白后荧光恢复实验(FRAP)证实了SLB膜具有连续有序结构.
采用传统滴涂法(drop-coating)制备MLB: 将150 µL体积的DOPC水溶液(0.5 mg/mL)直接滴在硅片表面, 置于真空环境中干燥. 溶剂蒸发后, 形成有序的多层磷脂膜. 样品在90%相对湿度下孵育0.5 h, 得到有序的多层结构. 在对照实验中通过FRAP和X射线衍射(XRD)测试证实了样品具有长程有序多层结构.
将样品加载至反应仓中纯水一侧(浸于溶液中的无脂膜覆盖区域事先以绝缘胶封闭); 另一侧加入一定体积的活性分子溶液, 确保混合后达到所需的溶液浓度. 通过模块化的功能性夹具设计, 保证了样品加载步骤中良好可重复性. 利用氙灯(500 W)对光电极进行方波调制照明(例如每个周期为50 ms ON和150 ms OFF), 收集输出电压信号; 在0时刻将纯水与反应液混合, 并同时记录输出电压信号随时间的变化.
-
在方波调制的光照下, 硅片中产生光致电荷并传递到电介质-磷脂膜上, 使膜液界面处的双电层带电(即充电); 当光照关闭时, 双电层放电(图2(a)). 在充放电过程中, 工作电极和参比电极之间的电压差经放大后被实时采集作为输出信号(表现为变形的方波). 需要注意的是, 由于双电层的厚度只有几纳米且非常靠近膜-溶液界面, 因此膜的结构和性质直接决定了双电层特性. 此外, 由于电路中电荷量小, 电流值小, 层中积累/剩余电荷量的变化可以表示为两电极间电压的变化. 因此每个光脉冲周期内所观察到的输出信号即反映了该时刻膜结构特性. 在活性分子与细胞膜相互作用的过程中, 实时采集输出信号(即双电层的充放电脉冲), 其变化将反映该过程中细胞膜结构性质的变化.
该系统可简化等效为图2(b)所示的电路, 其中可变电容
$ {C}_{1} $ 和可变电阻$ {R}_{1} $ 的并联表示工作电极(包括双电层)的贡献, 该贡献与膜特性相关; 电阻$ {R}_{2} $ 代表了其他部分(例如工作电极和参比电极之间溶液)的阻抗, 在单个测试系统中通常保持恒定. 由于半导体硅片的光电响应, 在方波调制光照射后(例如50 ms ON和150 ms OFF), 方波电压信号被输入该电路中. 电压波形经过等效电路后会发生变化, 形成输出波形, 通常为变形方波脉冲, 其形状直接取决于回路中$ {R}_{1} $ ,$ {R}_{2} $ ,$ {C}_{1} $ 的瞬时值(图2(c)).连续完整的DOPC SLB可以被视为电介质层, 其贡献可近似地表示为
$ {R}_{2} $ ,$ {R}_{1} $ 和$ {C}_{1} $ 的值分别为2 MΩ, 50 MΩ和50 nF. 基于等效电路和仿真, 可以绘制出图2(d)中的充放电曲线①. 然后, 通过拟合该轮廓的下降边缘, 能够定量获得弛豫时间常数τ (即$ {\tau }_{2} $ )的值(τ = 96.2 ms; 计算方法及含义详见3.2节). 如果活性分子吸附在膜表面上, 则会增大电路中$ {C}_{1} $ 的等效值, 因此, 可粗略假设$ {C}_{1} $ 值为80 nF, 并保持$ {R}_{2} $ 和$ {R}_{1} $ 的值不变, 将获得曲线②. 从这个新轮廓中可以得到一个新的τ值 (τ = 153.8 ms). 在活性分子作用下, 膜的结构会发生诸如渗透甚至破裂等变化, 导致$ {R}_{1} $ 的等效值突然下降, 可以假设其为较小值1 MΩ. 根据这一情况得到曲线③, 并从中获取了 τ 值(τ = 33.3 ms). 基于这些现象, 能够揭示膜结构变化与充放电曲线形态演变之间的关系(图2(e)). 换言之, 极性分子表面吸附通常对应着更长的τ值, 而膜透化则对应着τ值下降. -
已有3种方法被发展和应用于光电压瞬态实验定量和/或定性数据处理中, 他们分别适宜于不同的应用场景; 更多的数据处理方法, 例如利用小波变换分解充放电曲线中包含的不同频率过程信息, 目前正在研究中.
方法一 通过分析充放电曲线的弛豫时间常数, 可以定量描述其特性. 弛豫时间常数(τ)可从充电和放电过程曲线中计算得到. 对于光电压测试系统而言有
其中,
$ \bar{\tau } $ 为系统的等效电荷弛豫时间常数.$ \bar{\tau } $ 的值受半导体的性质(如厚度、电阻率、载流子迁移率等)和溶液(如溶质、浓度、扩散系数等)的影响. 在此处, 分别引入$ {\tau }_{1} $ 和$ {\tau }_{2} $ 来表示半导体和溶液的影响. 其中, 半导体材料的电荷弛豫时间常数($ {\tau }_{1} $ )对于特定类型光电极而言是一个固定值, 在本系统中保持不变; 而溶液的电荷弛豫时间($ {\tau }_{2} $ )则会显著影响整个等效电荷弛豫时间($ \bar{\tau } $ ). 当$ {\tau }_{2} $ 的贡献远大于$ {\tau }_{1} $ 时,$ {\tau }_{2} $ 的变化将直接反映膜性质的变化.在简化等效电路中, 充电和放电过程皆可用于定量化分析. 方便起见, 以放电过程为例:
式中
$ q\left(t\right)={C}_{1}\varepsilon {{\mathrm{e}}}^{-t/R{C}_{1}} $ , 其中R为从$ {C}_{1} $ 位置获得的电路等效电阻($ \dfrac{{R}_{1}{R}_{2}}{{R}_{1}+{R}_{2}} $ ), ε为光激活半导体产生的电势,$ R{C}_{1} $ 的乘积即为τ.当时间t = τ, Q(t)(和U(t))的数值等于最大值的
$ {{\mathrm{e}}}^{-1} $ . 因此, 可以近似地将电压脉冲下降沿(即放电过程)电压值下降$ {1-{\mathrm{e}}}^{-1} $ ≈70%的时间作为τ值来定量刻画该放电过程(如图3(a), (b)中红色部分所示). 简而言之, 采集充放电脉冲下降沿电压值下降70%的时间作为τ, 绘制τ-t曲线并加以分析, 这是该数据处理方法的核心. 通过τ随时间变化的测量, 可以获得膜结构性质随时间动态改变的信息, 例如待测膜的水化/去水化、活性分子在膜表面的吸附/解吸附、膜结构的扰动和破坏; 尤其是, τ的变化时间点提供了不同作用状态之间转变的时间信息. 这些信息的获取于大多数传统技术而言具有挑战性. 因此, 将该技术与传统技术结合使用, 能获得良好效果(关于膜结构与τ之间定量关系的数值仿真结果见图2(d), (e); 实际应用举例详见4.1和4.2节).方法二 最大电压值分析. 基于(2)式和(3)式, 在保持硅片基底和光照条件不变的情况下, 单脉冲内的最大电压值
$ {U}_{{\mathrm{m}}{\mathrm{a}}{\mathrm{x}}} $ 与等效电容$ {C}_{1} $ 、电阻$ {R}_{1} $ 之间存在定量关系, 如仿真图2(e)所示. 因此,$ {U}_{{\mathrm{m}}{\mathrm{a}}{\mathrm{x}}} $ 随时间的变化可反映膜结构的演化过程(例如图3(c)展示了多层磷脂膜与蜂毒肽相互作用中$ {U}_{{\mathrm{m}}{\mathrm{a}}{\mathrm{x}}} $ 随时间的变化). 特别是在分析跨膜孔形成等现象时, 即使是对于膜通透性改变这样复杂的过程, 也可以简便有效地利用基于Umax的分析方法进行研究, 并在无需标记和第三方检测分子的情况下提供膜透化现象发生的初始时刻信息(表现为$ {U}_{{\mathrm{m}}{\mathrm{a}}{\mathrm{x}}} $ 的骤降, 具体应用案例请参见4.4节). 相比 之下, 在传统技术中常用于检测膜通透性的方法包括巨囊泡泄漏动力学检测和电化学检测, 前者需要额外添加第三方荧光分子, 根据荧光分子是否发生跨膜扩散来判断膜通透性的变化, 后者则需要施 加额外电场. 这些因素都有可能影响待测生物学 过程.方法三 过充电情况分析. 在弱负电荷CDs与磷脂膜相互作用的过程中, 观察到了膜的过充电和过放电现象(图3(d)). 这种现象可以归因于衬底和嵌入在SLB中的导电碳点之间发生了电荷转移. 尽管潜在机制尚不明确, 但这些结果证实了碳点对脂质膜结构和电性能产生干扰作用(具体细节请参见4.5节). 光电压瞬态技术在不需要施加外加偏压的情况下提供待测薄膜的电学性质信息, 这于传统技术而言是一个有益的补充(例如常用于实时监测界面分子相互作用过程的带耗散的石英晶体微天平技术(QCM-D)尽管也可以追踪界面分子质量和能量耗散情况随时间的动态变化过程, 但是无法提供电学性质相关的信息).
-
由于活细胞膜在组分、结构异质性等方面的复杂性以及易受膜内外环境影响等特点, 模型细胞膜, 特别是支撑脂质单层膜和多层膜被广泛用于细胞膜界面相关的研究中. DOPC是最常用的模型细胞膜脂质成分之一. 我们首先利用纯DOPC分子, 在硅片基底上构建了干燥的SLB和MLB薄膜, 并获取了其在光电压瞬态分析中典型的充放电曲线, 进而监测了它们在纯水中的水合溶胀过程[38]. 以往工作通过XRD和QCM-D技术实时监测单层和多层磷脂膜的溶胀过程, 提供Z方向层状结构厚度和薄膜质量变化信息; 而光电压瞬态技术则进一步提供溶胀过程动力学信息以及不同阶段之间发生转变的时间信息. 该工作证明了光电压瞬态技术在实时监测膜界面过程中的有效性; 尤其是, 定量测量了DOPC磷脂膜在光电压瞬态测量中的特征τ值, 为后期进一步发展对工作电极薄膜结构的定量分析方法提供了可能.
图4(a)展示了将干燥的纯DOPC单层膜暴露于纯水中时, 其光电压充放电曲线及τ值随时间演化的过程. 在整个实验过程中, τ值基本保持恒定(4.8 ± 0.4) ms, 该数值可视为DOPC单层膜的特征τ值, 表明SLB膜在水浸和光电测量过程中保持相对稳定且未发生明显变化或损坏. 室内XRD测量结果在不同时刻验证了这一结论(图4(a3)). 然而, DOPC多层膜(厚度约为350 nm)呈现出不同现象(图4(b)), 其τ值从初始的4.8 ms迅速增至约12 ms (大约5 min时刻), 之后保持短时间动态平衡, 并降至约8 ms (10—15 min时刻), 最终处于强波动的动态平衡状态(>15 min), τ值在 (7.7 ± 1.2) ms. 这些现象反映了多层膜逐渐溶胀并最终达到饱和状态. 当暴露于水中时, 极性水分子逐渐渗透到MLB亲水头基层之间, 增强了等效电容, 导致较大的τ值; 然而随着更多水分子到达膜-硅界面, 等效电阻
$ {R}_{1} $ 急剧下降, 导致τ值迅速减小; 之后, 整个系统基本达到平衡, 并伴随着多层膜内轻微的溶胀和/或脂质重组. 这些动态过程在传统XRD分析中难以体现(图4(b2)). 需要注意的是, 在SLB (干膜/水合状态)和MLB (干膜状态)两种情况下, 它们都具有相似的特征τ值. -
由于细胞膜脂质分子的两亲性结构特征, 表面活性剂(尤其是阳离子表面活性剂)与脂质相互作用研究对新型纳米药物或纳米载体的开发具有重要指导意义. 大量传统方法被应用于相关研究中, 包括AFM (表征膜表面形貌在活性分子作用前后的变化)、QCM-D (实时监测活性分子作用下膜的质量变化和能量耗散情况改变). 一些其他技术, 例如超分辨荧光成像、单分子荧光共振能量转移、核磁共振等, 则可以提供高空间分辨率甚至分子层次的分子间相互作用信息. 我们通过将光电压瞬态技术与传统方法(巨囊泡泄漏监测技术、AFM和分子动力学模拟)相结合, 实时监测了具有代表性 的阳离子表面活性剂-十四烷基三甲基溴化铵(TTAB)在宽浓度范围(0.5 μmol/L—10 mmol/L)下与磷脂双层膜的相互作用动态过程, 补充揭示了不同作用状态的存在, 并提供了膜在不同作用状态之间转变的时间信息(图5)[39]. 结果显示, 在低浓度条件下(例如≤10 μmol/L), TTAB的作用过程可分为3个阶段, 包括结构扰动主导阶段(体现 为τ随时间降低, 图5(b1)—(b3), Stage I)、吸附主导阶段(体现为τ显著增大, Stage II)和动态平衡阶段(体现为τ波动, Stage III). 随着TTAB浓度增大, 该过程被加速. 在较高浓度条件下(例如≥100 μmol/L), 该过程缩减至两个阶段(图5(e1), (e2)). 此外, 由于表面活性剂分子在双分子层上引起不对称扰动, 在较高浓度情况下TTAB会导致膜变形甚至破裂. 光电压瞬态监测将膜作为整体, 提供了这些转变发生的时间点. 该工作证明了光电压瞬态技术能够作为传统技术的有效补充, 简单、实时地探索膜界面上的动态过程.
-
聚合物分子作为纳米药物载体材料具有重要意义, 在临床应用中, 其与细胞膜之间的动态相互作用不容忽视. 以两种典型聚合物分子——聚氧乙烯十二烷基醚(Brij35)和聚乙烯吡咯烷酮(PVPk30)为例, 我们继而对聚合物分子与磷脂膜的相互作用过程进行了探究[40]. 结果发现, 调节溶液中聚合物浓度, Brij35主要表现出吸附-积累-渗透的过程; 而相比之下, PVPk30在分子吸附-解吸附和/或膜渗透-愈合等竞争性机制之间呈现动态平衡. 这种在膜作用过程中的差异解释了它们在活细胞实验中所表现出高低细胞毒性的差别. 通过数据点波动反映竞争性机制共存, 这是光电压瞬态技术的优势之一, 源于其将整个膜作为研究对象以及高灵敏度特点.
-
多肽以及基于多肽的药物设计为解决严重疾病或感染问题提供了有希望的解决方案. 抗菌肽(antimicrobial peptide, AMP)和细胞穿膜肽(cell penetrating peptide, CPP)是两类典型的膜活性多肽, 它们通过与细胞膜相互作用来发挥生物学功能. 以AMPs为例, 其通常具有带正电、亲疏水并存的双亲结构, 在细菌细胞膜上形成孔洞或引起膜水解、导致透化和细菌内容物泄漏从而杀灭细菌. 基于天然抗菌肽进行序列改造或仿生设计等方法开发高效率破膜分子是获取新型抗菌药物的重要途径. 多肽(及多肽类药物)典型的膜作用机制包括多肽在膜表面的吸附、累积以及插入导致跨膜缺陷和水解等过程, 同时, 某些多肽还表现出反复插入-抽出-再插入的动态过程, 这些机制特别适合利用光电压瞬态技术来追踪和表征. 因此, 该技术为实时监测多肽对磷脂膜的不同扰动机制提供了简便途径; 尤其是, 它能够揭示不同作用机制发生转变的时间信息, 这对其他大多数传统技术而言是一个挑战.
首先, 该技术被用于揭示多肽作用下磷脂膜出现跨膜孔的时间. 以蜂毒肽(melittin)和聚乙二醇(PEG)为基础, 设计了一系列不同构象和抗菌效能复合物; 其中, 将单个melittin末端共价连接到长链PEG上得到的PEG12k-1*Mel分子表现出最佳抗菌效果, 其抗菌效率比原始melittin分子提高了5倍(图6(a), (c))[41]. 光电压瞬态技术被应用于评估melittin和PEG12k-1*Mel分子对脂质双层透化能力; 在与SLB相互作用过程中, 实时记录了充放电曲线中最大电压值Umax. 如图6(b)所示, 在没有药物的情况下, Umax随时间保持不变, 表明膜-液界面处于稳定状态. 添加药物后, Umax呈现3个阶段的变化: 指数下降、突然下降和较慢的指数下降, 并最终达到平衡. Umax的变化反映了药物与膜之间的动态相互作用, 包括药物在膜表面吸附和积累(导致膜变薄和延展)以及跨膜缺陷形成和膜界面分子重组; 其中, Umax骤降时刻对应于跨膜缺陷出现时间. 实验结果显示, 在t = 10 min时纯melittin(浓度为5 μg/mL)发生电压骤降, 而PEG12k-1*Mel大大缩短了该时间, 即使在更低的浓度下, 其破膜速率也远高于纯melittin (例如0.5 μg/mL浓度下, Umax发生骤降的时间为2 min). 这些结果展示了PEG12k-1*Mel相较于melittin更高的破膜效率.
由于氨基酸序列和结构的多样性, 与AMPs相比, CPPs的膜作用机制更为复杂(图7). 我们进一步利用光电压瞬态技术结合巨囊泡泄漏动力学等实验研究了TAT多肽(序列为YGRKKRRQRRR)与模型细胞膜之间的相互作用过程[44]. 在不同多肽浓度下, 不同反应体系中弛豫时间常数τ随时间发生显著波动, 揭示了某些竞争性过程存在(例如膜去水化-水化、多肽吸附-解吸附、膜透化-愈合). 此外, 在1.0 μg/mL浓度下, 多肽引起τ值增大(t ~ 1 min)、波动(1—10 min)、降低(t ~ 10 min), 最终达到小于初始值的动态平衡状态(>10 min), 分别对应着多肽在膜表面吸附、TAT-脂质复合结构的重构、以及膜完整性的严重破坏. 当浓度为10.0 μg/mL时, 该过程被加速: 多肽添加立即导致τ减小和膜完整性破坏, 并使体系最终τ值远低于初始值. AFM表征验证了TAT多肽对膜结构的扰动.
多肽分子间的协作是突破能量势垒、实现从膜表面吸附到跨膜插入的重要原因. 此外, 不同种类分子之间广泛存在着协同作用. 多肽PGLa和Magainin 2 (简称MAG2)之间的协同作用为提高其抗菌能力提供了有效途径, 然而其协作机制尚未完全阐明. 我们将界面光电压测试与巨囊泡泄漏实验和分子动力学模拟相结合, 对PGLa, MAG2以及PGLa/MAG2混合物的动态膜穿孔过程进行研究[42]. 研究发现这3个系统遵循不同的膜相互作用模式, 特别是PGLa-MAG2异二聚体形成对于其高效破膜成孔功能至关重要. 尽管PGLa和MAG2都含有相似数量的疏水性残基和正电荷残基, 但它们在相互协作中扮演着不同角色: PGLa易于插入膜中, 而MAG2则以膜结合构象进一步稳定其产生的缺陷/孔. 此外, MAG2负责招募多肽以形成PGLa-MAG2异二聚体甚至组装异二聚体簇, 并最终导致孔道逐渐增大并引起跨膜渗漏. 该研究深入揭示了在形成膜孔过程中PGLa和MAG2之间复杂的协同机制, 同时也证明了光电压瞬态技术与传统技术相结合能够补充提供多肽-膜相互作用过程的动力学信息和时间信息.
-
除了非导电材料外, 导电材料(例如纳米颗粒)对膜电学性质的影响更能被光电压瞬态技术灵敏捕捉, 这对于QCM-D、光学成像及光谱分析等大多数传统界面技术而言是个挑战. 小尺寸荧光CDs在生物医学领域的应用备受关注. 带正电CDs的环境和生物相容性已经得到广泛研究; 然而, 对于带负电或电中性的CDs通常被认为是无生物毒性的观点(即使在较高浓度下), 需要重新评估. 我们首先通过结合巨囊泡泄漏动力学、QCM-D、同步辐射XRD、分子动力学模拟等多种生物物理方法, 系统地研究了直径为3 nm且具有弱负电荷特性的CDs对细胞膜结构所产生的破坏作用[45]. 研究结果表明, 即使在0.5 μg/mL的低浓度下, CDs也会对细胞膜产生显著的结构扰动, 导致脂膜双层结构不对称地被扰乱从而增加了膜通透性. 此外, 在不同浓度条件下, CDs展现出不同机制: 在低浓度(<20 μg/mL)时迅速插入双分子层, 并在高浓度(例如25—200 μg/mL)时呈现颗粒间协同效应. 图8(b), (c)分别展示了使用QCM-D和XRD测试发现的碳点导致脂膜双层结构不对称破坏和层内脂分子间距增大的情况.
之后, 光电压测试被用于分析CDs对磷脂膜电学性质的影响(图8(d)). 实验结果显示, 低浓度CDs(例如≤5 μg/mL)对脂膜的影响较为微弱; 然而, 当CDs浓度达到15 μg/mL时, 脂膜经历了明显的过充电和过放电现象(图8(d3)). 这种现象在CDs浓度升至50 μg/mL时更加显著(图8(d4)). 参考实验中以裸硅片为基底(无SLB)加入CDs后并未观察到类似现象, 这表明导致过充电现象的原因是由于CDs与双层膜之间发生相互作用. 推测过充电(和过放电)现象可能源自硅基底和嵌入在SLB中具有导电性质的CDs之间存在着电荷转移. 尽管其具体机制尚不清楚, 但通过光电压实验结果证实了CDs对于脂质膜结构和电性能所产生的干扰作用.
-
光电压瞬态技术为固液界面上发生的动态过程提供了一种原位、实时、整体、非侵入、无需标记以及高灵敏度的实验手段, 具备较高时间分辨率(百毫秒级). 以活性分子与人工细胞膜相互作用研究为例, 该技术能够揭示活性分子诱导磷脂膜结构和性质变化的动力学过程, 并提供不同状态之间转变的时间信息. 然而, 目前仍存在一些待解决的问题和缺点.
首先, 目前光电压瞬态设备的信号采集原理依赖于体系中仅存在较小的电流(<1 nA), 故而要求待测过程在低离子溶液中进行. 因此, 生物体系中常见的溶液环境(例如PBS缓冲液、细胞培养基等)在目前的设备体系中不适用. 该问题的解决, 有待于设备中信号采集模块的改造.
其次, 输出信号的充放电曲线在揭示待测体系的分子相互作用中具有非特异性. 这体现在两个方面. 首先, 目前实验结果能够有效区分导电纳米颗粒和聚合物、多肽等活性分子, 原因是前者表现出硅基底和纳米颗粒之间发生电荷转移引起的过充电和过放电现象; 然而, 对于聚合物、多肽、表面活性剂等活性分子, 在充放电曲线上并未观察到明显差异, 在光电压实验结果中的差异体现了它们不同的膜作用过程. 其次, 仅依靠某一时刻的充放电曲线来表征膜系统即时结构特性(如分子填充状态), 或者仅依赖时间相关的τ分布来确定未知化合物系统动态相互作用分子机制都是具有挑战性的. 为了克服这些局限性, 需要有效结合其他研究方法. 例如, 传统生物物理实验技术(如巨囊泡泄漏动力学监测、QCM-D等)能够提供关于活性分子的膜作用机制的相关认识, 从而为光电压实验现象提供合理解释的理论基础; 后者进一步补充了包括相转变时间在内的信息. 同时, AFM、XRD和计算机模拟等手段可以提供待测体系表面形貌和内部结构的相关信息[45–47]. 总之, 多种实验和模拟手段的有效结合是研究生物界面时间空间多尺度复杂过程的途径.
同时, 由于测试系统的敏感性, 目前的光电压瞬态设备对工作电极的质量提出了较高的要求. 例如, 硅片质量、表面清洁效果、硅片与磷脂膜之间贴附均匀性以及磷脂膜的完整性等因素都会对输出信号的定量数值产生影响.
当前光电压瞬态技术仍处于初步阶段, 可作为现有界面技术的有益补充. 本文主要总结了该技术在支撑磷脂膜与膜活性分子之间相互作用领域的应用. 此外, 在其他界面体系中, 如真实细胞膜的界面生物过程追踪和基底表面吸附分子与溶液内活性分子之间相互作用研究等方面, 该技术也具备提供有益信息的潜力. 目前我们正专注于改进光电极样品制备的可重复性和测量稳定性, 并扩展待测体系范围. 另外, 我们还致力于建立基于膜模型重构的充放电曲线定量分析模型, 并利用小波变换等方法深入挖掘数据信息. 随着该技术进一步发展, 它在定量分析、天然细胞膜生物过程分析以及分子间动态相互作用追踪和生物传感器等领域应用有望在不久的将来得以实现.
光电压瞬态技术: 实时分析膜界面动态过程的新手段
Photovoltage transient technique: an alternative approach to real-time analysis of membrane interface dynamic processes
-
摘要: 活性分子与细胞膜之间的相互作用在许多基本的生物过程中扮演着至关重要的角色, 然而如何实现对此界面动力学过程的原位、实时、无标记且无侵入监测仍是生物物理研究领域所面临的一大挑战. 我们与合作者开发的光电压瞬态技术, 为解决这一问题提供了一种新途径. 该技术利用硅片光电响应生成电荷, 并将磷脂膜的充放电过程记录为电压瞬态脉冲、建立了该充放电过程与界面瞬时结构和性质之间的关联性. 因此, 通过对随时间演化的电压脉冲进行分析, 可以揭示活性分子作用下膜结构实时动态变化情况, 尤其是不同作用状态之间转换的时间信息, 可作为传统技术的有益补充. 同时, 该技术设备搭建成本低廉, 操作方便, 无需复杂的数据处理过程. 本综述概述了光电压瞬态技术的工作原理、设备搭建以及数据处理方法, 并以经典细胞膜模型——磷脂双层膜为例, 总结了该技术在探索磷脂膜水合特性及其与活性分子(如表面活性剂、聚合物、多肽和纳米颗粒)相互作用机制方面取得的最新进展. 最后就该技术优缺点进行讨论并展望未来发展前景.Abstract:
The dynamic interactions between active molecules and the cell membrane play a crucial role in various fundamental biological processes. The photovoltage transient technique developed in recent years has provided an in situ, real-time, and non-invasive approach to studying dynamic processes at the membrane interface. In this technique, the photoelectric response of silicon wafers is used to generate charges and record voltage transient pulses in the charging and discharging process of phospholipid membranes. These pulses directly reflect the instantaneous structure and properties of the membrane. By analyzing the temporal evolution of voltage pulses, the dynamic changes in membrane structure induced by molecular actions can be elucidated. In particular, this technique provides valuable insights into the timing of transitions between different functional states. In this paper, the working principle, equipment setup, and data processing methods used in photovoltage transient analysis are comprehensively reviewed. Furthermore, the use of supported phospholipid bilayers as model cell membranes highlights recent progress of this technique in investigating the mechanisms underlying membrane interactions of active molecules such as surfactants, polymers, peptides, and nanoparticles. Finally, its advantages and limitations are evaluated, and its development prospects are discussed. The photovoltage transient technique is initially used to analyze the charging and discharging curves, as well as the hydration process, of single- and multi-layered membranes composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) phospholipids. Previously, X-ray diffraction (XRD) and quartz crystal microbalance with dissipation (QCM-D) technology were commonly utilized for the real-time monitoring of the swelling process in phospholipid membranes, providing the information about changes in mass and thickness of Z-direction layers. In contrast, the photovoltage transient technique presents additional insights into the kinetics of the swelling process and timing of transitions between different stages. The present study demonstrates the effectiveness of photovoltage transient technique in real-time monitoring of membrane interface processes, specifically, it quantitatively measures the characteristic τ value of DOPC phospholipid membrane, thereby enabling further development of quantitative analysis method for this technique. Then, the photovoltage transient technique, in conjunction with giant unilamellar vesicle (GUV) leakage assays, atomic force microscopy (AFM) and QCM-D, is employed to monitor the structural perturbation of surfactants (TTAB) and polymers (Brij35 and PVPk30) on the membranes. Specifically, Brij35 primarily undergoes an adsorption-accumulation-penetration process; whereas PVPk30 exhibits a dynamic equilibrium between molecular adsorption-desorption and/or membrane permeation-healing competing mechanisms. This disparity in membrane action processes elucidates the discrepancy observed in their cytotoxicity during live cell experiments. The ability of photovoltage transient technology to investigate the entire membrane as a research subject along with its high sensitivity enables it to capture fluctuations in data points that reflect the coexistence of competitive mechanisms. Furthermore, photovoltage monitoring reveals the occurrence of peptide-induced membrane permeabilization. The distinct mechanism of action on the membrane between melittin (as a representative antimicrobial peptide) and TAT (a typical cell penetrating peptide) is elucidated. Finally, the conductive carbon dots (CDs) induced phenomena of membrane overcharging and overdischarging, are likely to be attributed to charge transfer between the silicon substrate and the embedded conductive CDs. -

-
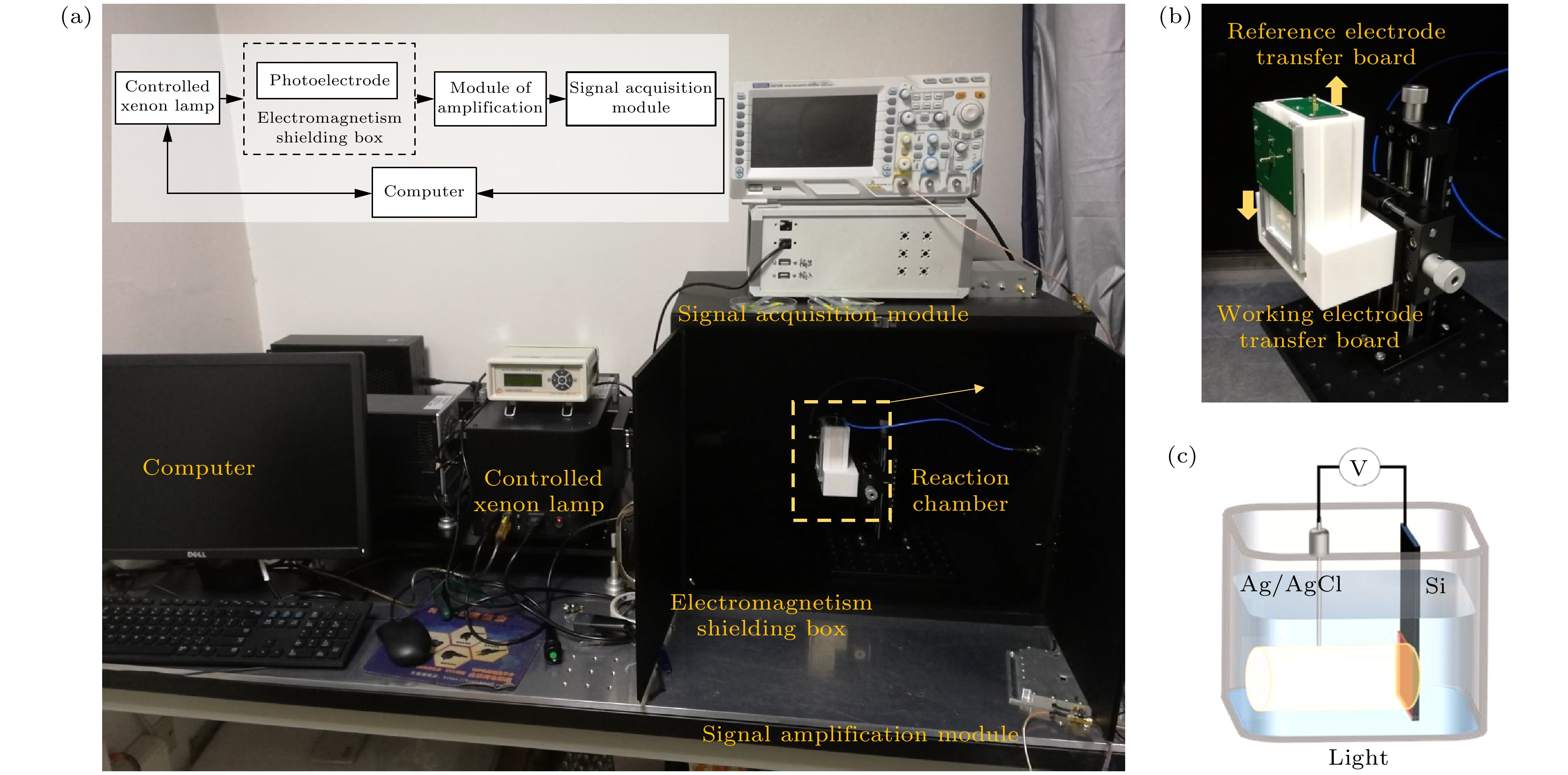
图 1 光电压测试系统和反应仓的照片及示意图 (a) 设备照片, 该套系统包含置于电磁屏蔽箱内的反应仓(内含工作电极和 参比电极)、信号放大模块、信号采集模块、可控光源和计算机, 其中插图为对应的模块分布示意图; (b) 反应仓照片, 功能夹具 可实现方便和稳定的样品安装和实验测量; (c) 反应仓示意图, 反应仓由浸泡于活性分子溶液中的硅片及其表面覆盖的磷脂膜(无膜部分以绝缘胶覆盖)、参比电极、方波调制的光照组成, 工作电极与参比电极之间的电压差被实时采集; 部分图片引用和 修改自文献[38]
Figure 1. Photos and schematic diagrams of the photovoltaic testing system and reaction chamber. (a) Photo of the experimental setup. The system includes a reaction chamber (containing working electrodes and reference electrodes) placed inside an electromagnetic shielding box, a signal amplification module, a signal acquisition module, a controllable light source, and a computer. Inset shows simplified settlement of the system. (b) Photo of the reaction chamber. The functional fixture can achieve convenient and stable sample installation and experimental measurement. (c) Schematic diagram of the reaction chamber, which consists of a silicon wafer immersed in an active molecule solution and its surface covered with a phospholipid membrane (the non membrane part is covered with insulating glue), a reference electrode, and square wave modulated light. The voltage difference between the working electrode and the reference electrode is collected in real time. Images are cited and modified from Ref. [38].
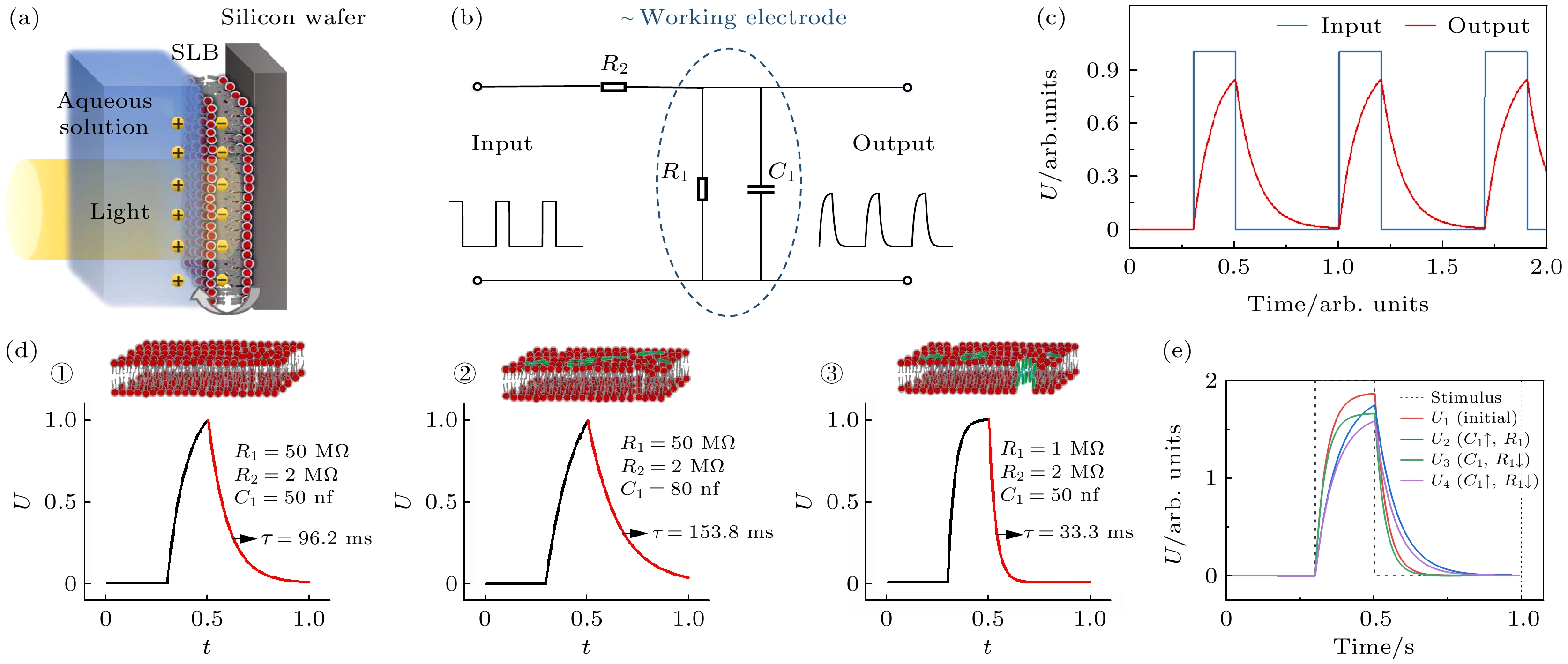
图 2 光电压测试系统的简化等效电路及数值仿真 (a) 工作电极示意图, 硅片基底在方波调制光照下发生光电响应和生成电荷, 导致膜液界面双电层发生充放电; (b) 简化等效电路, 其中, 可变电阻
$ {R}_{1} $ 与可变电容$ {C}_{1} $ 的并联等效于工作电极的贡献; (c) 输入信号与输出信号的典型仿真曲线, 分别为方波信号和变形方波信号; (d) 膜结构改变与输出电压曲线之间的关联,$ {R}_{1} $ ,$ {R}_{2} $ ,$ {C}_{1} $ 取假设数值、对应于不同膜结构, 曲线为基于等效电路的仿真结果, τ为基于曲线计算出的弛豫时间常数; (e) 膜结构变化(即$ {R}_{1} $ ,$ {C}_{1} $ 数值改变)导致的充放电曲线对比. 部分图片引用和修改自文献[38]Figure 2. Simplified equivalent circuit and numerical simulation of photovoltage testing system. (a) Schematic diagram of the working electrode. The silicon wafer substrate undergoes photoelectric response and generates charges under square wave modulation light, resulting in charging and discharging of the double layer at the film liquid interface. (b) Equivalent circuit of the test system, where the parallel connection of variable resistor R1 and variable capacitor C1 is equivalent to the contribution of the working electrode. (c) Representative simulation profiles of the input (in blue) and output (red) signals, they are square wave signal and deformed square wave signal, respectively. (d) Relationship between changes in membrane structure and changes in the output profile. The input postulated values of
$ {R}_{2} $ ,$ {R}_{1} $ and$ {C}_{1} $ , as well as the as-calculated τ value based on the circuit in (b) are shown in each condition. (e) Comparison between photo-voltage pulses due to changes in$ {R}_{1} $ and$ {C}_{1} $ values. Images are cited and modified from Ref. [38].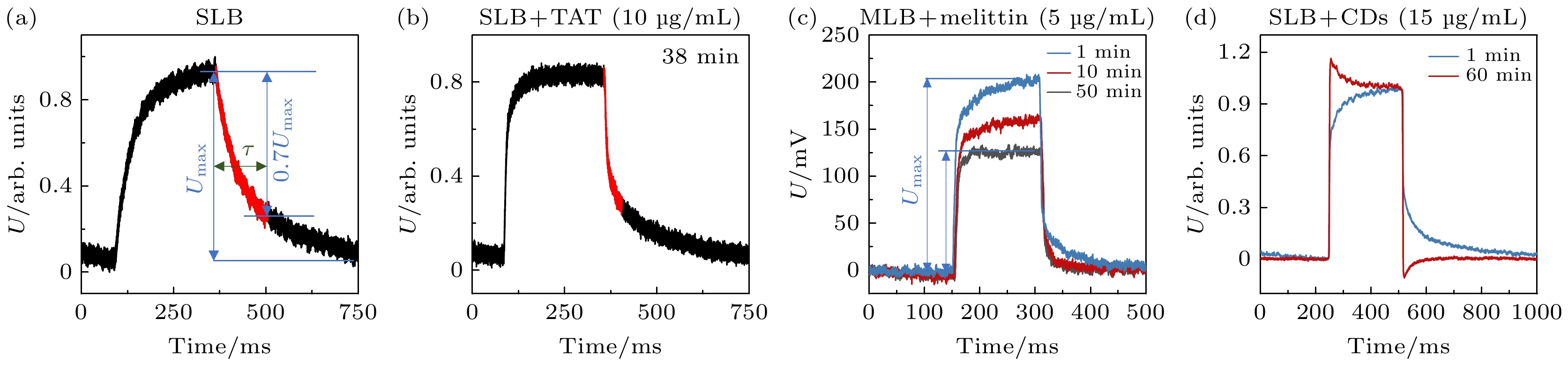
图 3 光电压瞬态技术中的数据采集与分析方法举例 (a) 纯DOPC单层磷脂膜的典型充放电曲线; (b) 加入TAT多肽38 min后, 体系的充放电曲线; (c) 纯DOPC多层膜(MLB)与蜂毒肽相互作用不同时刻的体系充放电曲线; (d) 纯DOPC单层膜与碳点(carbon dots, CDs)相互作用不同时刻的体系充放电曲线. (a)—(d)皆显示了一个脉冲的典型曲线. 脉冲的时间长度取决于实验设定的光脉冲ON、OFF时长, 不影响对τ值的计算. (a), (b), (d)中展示了电压经归一化处理之后的结果; (c)显示了未经归一化处理的原始曲线. 图片引用和修改自文献[44,45]
Figure 3. Examples of data acquisition and analysis methods in photovoltage transient technology: (a) The charge and discharge curve of a pure DOPC SLB, voltage is normalized; (b) the charge and discharge curve of the system after adding TAT peptide for 38 minutes, voltage is normalized; (c) the charge and discharge curves of pure DOPC MLB with melittin at different time points of interaction, voltage is not normalized; (d) the charge and discharge curves of pure DOPC SLB interacting with carbon dots (CDs) at different time intervals, voltage is normalized. Panels (a)–(d) all exhibit typical curves of pulses, and the duration of the pulses depends on the experimentally set ON and OFF duration of the light pulses, which does not affect the calculation of the value of τ images are cited and modified from Ref. [44,45].
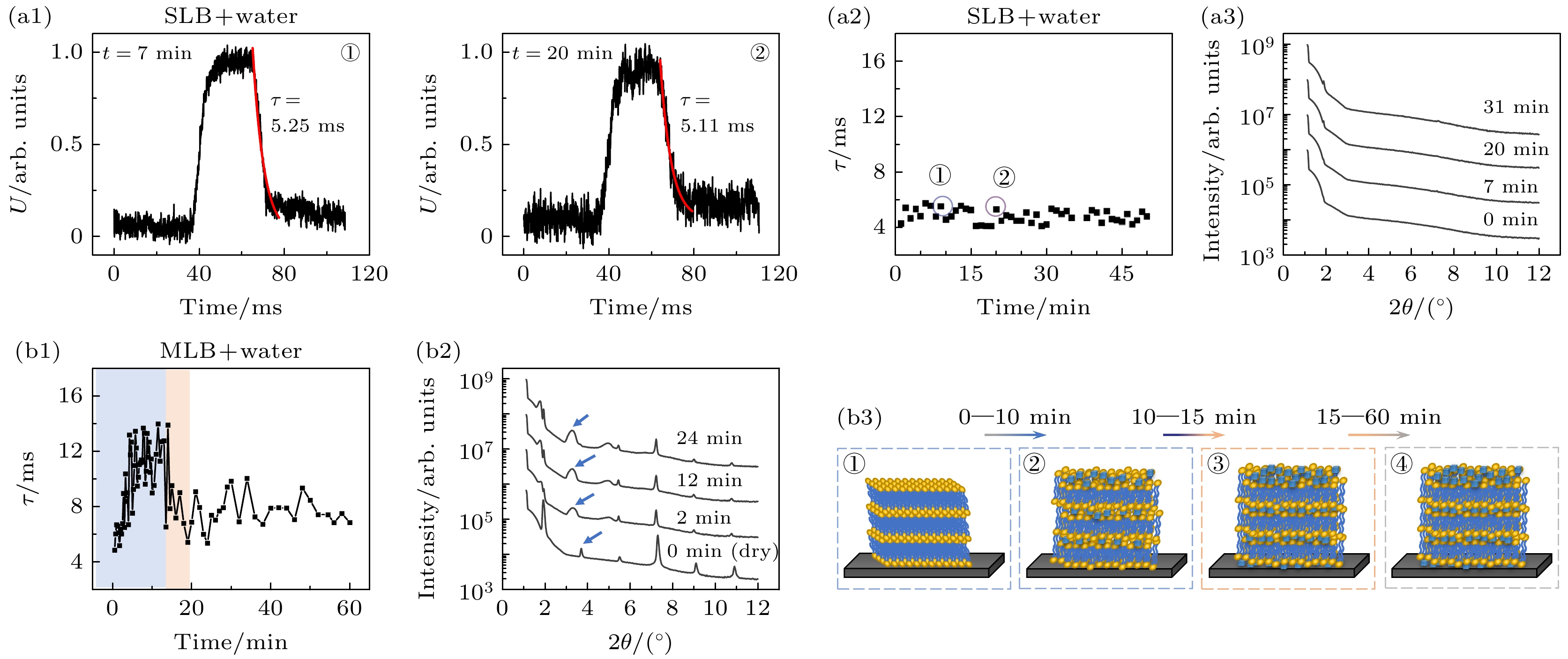
图 4 纯DOPC单层膜(a1)—(a3)和多层膜(b1)—(b3)在光电压瞬态分析中的典型充放电曲线及其水合过程 (a)单层膜在不同时刻的充放电曲线、水合过程中τ值随时间的演变、及不同时刻的室内XRD曲线(典型测量结果); (b)多层膜在水合过程中τ值随时间的演变、不同时刻的室内XRD曲线、及对应的水合过程示意图, 蓝色珠子代表水分子, 其中(b1), (b2)为典型测量结果. 图片引用和修改自文献[38]
Figure 4. Typical charge-discharge curves and hydration processes of pure DOPC SLB (a1)–(a3) and MLB (b1)–(b3) membranes in photovoltage transient analysis. (a) The charge and discharge curves of a SLB at different time points, along with the evolution of τ value during hydration and XRD curve at various time intervals (typical measurement results). (b) The evolution of τ value over time, indoor XRD curves at different time points, and corresponding hydration process diagram for the MLB. The blue beads represent water molecules. (b1), (b2) are typical measurement results. Images are cited and modified from Ref. [38].
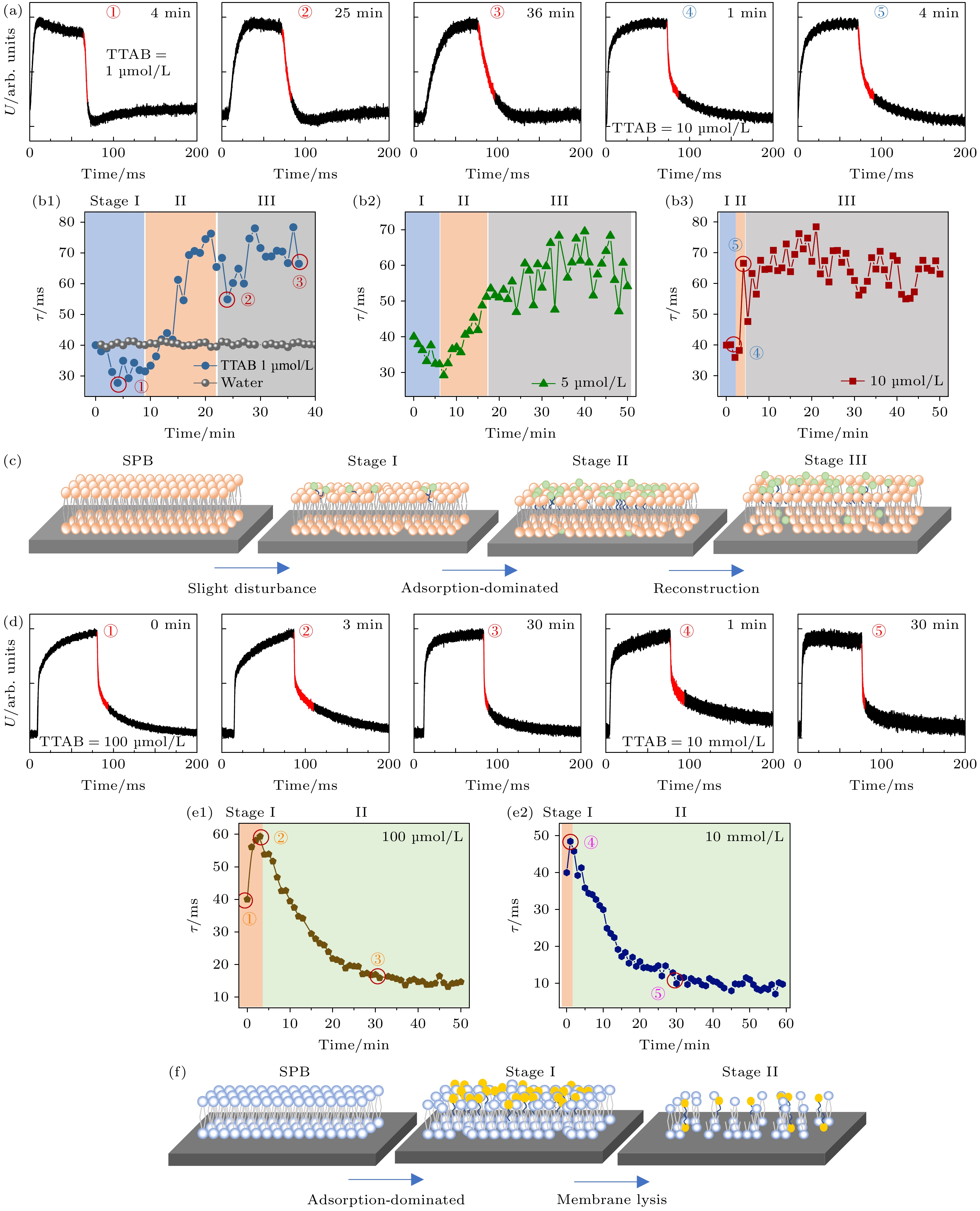
图 5 光电压瞬态技术分析不同浓度TTAB与DOPC磷脂单层膜相互作用的动态过程 (a), (d)典型的光电压脉冲曲线, 其对应的采集时刻以圆圈标于(b), (e)中; (b), (e) TTAB与磷脂膜相互作用过程中τ随时间演化的典型曲线, 体系初始τ值被归一化至40 ms, TTAB浓度分别为1, 5, 10, 100 μmol/L和10 mmol/L, 不同的作用阶段以不同背景色标出; (c), (f) 不同阶段中TTAB与脂质相互作用机制示意图. 图片引用和修改自文献[39]
Figure 5. Dynamic interaction between TTAB and a DOPC SLB with varying concentrations under the photovoltage transient analysis. (a), (d) Typical photovoltage pulse curves, with the corresponding acquisition time circled in (b), (e). (b), (e) Typical time evolution of τ during the interaction between TTAB and a DOPC membrane. The initial value in each test was normalized to 40 ms. The concentrations of TTAB used were 1, 5, 10, 100 μmol/L and 10 mmol/L respectively. Different stages of action are indicated by different background colors. (c), (f) Schematic diagrams illustrating the interaction process between TTAB and lipids. Images are cited and modified from Ref. [39].
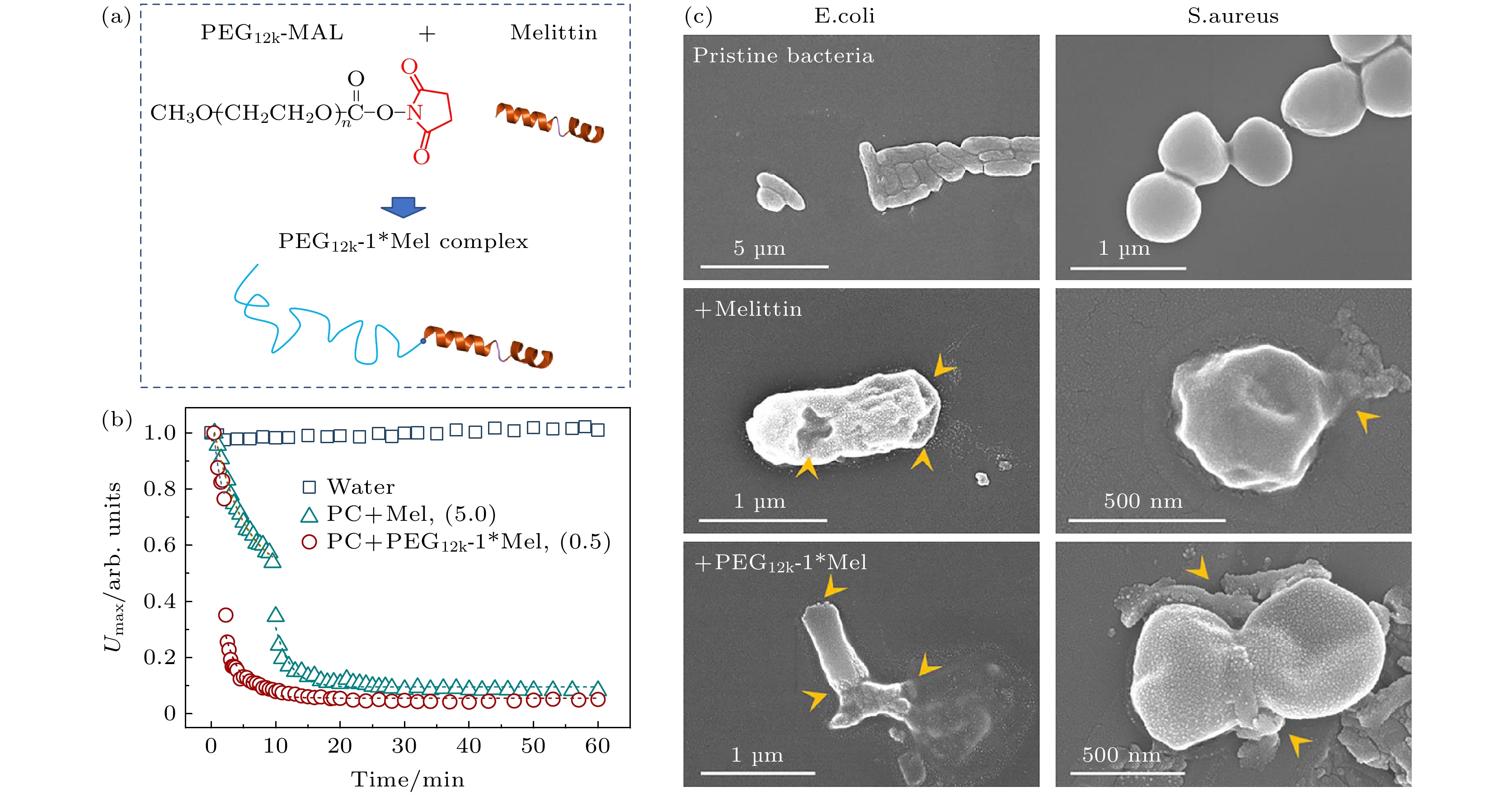
图 6 蜂毒肽(melittin)及其复合物(PEG12k-1*Mel)的膜作用活性分析 (a) PEG12k-1*Mel的制备及分子构象示意图; (b)光电压瞬态技术检测两种分子与SLB (成分为纯DOPC)的相互作用过程中Umax随时间的演变, 在0时刻加入药物分子, 浓度分别为5.0和0.5 μg/mL (以分子内多肽质量为标准来标定复合物浓度); (c)两种分子杀菌效果的SEM照片(药物浓度皆为64 μg/mL, 孵育时间16 h). 箭头标出了细菌膜破裂的位置. 图片引用和修改自文献[41]
Figure 6. Membrane activity of melittin and PEG12k-1*Mel: (a) Conjugation route of the PEG12k-1*Mel complex; (b) typical Umax-t profiles of DOPC membranes exposed to DI water, pure melittin (5.0 µg/mL) or PEG12k-1*Mel (0.5 µg/mL); (c) representative SEM images of E. coli and S. aureus bacteria after 16 h exposure to native melittin, PEG12k-1*Mel (peptide concentration of 64 µg/mL (in respect to peptide)), or no agent, where arrowheads indicate collapses of bacterial membrane. Images are cited and modified from Ref. [41].
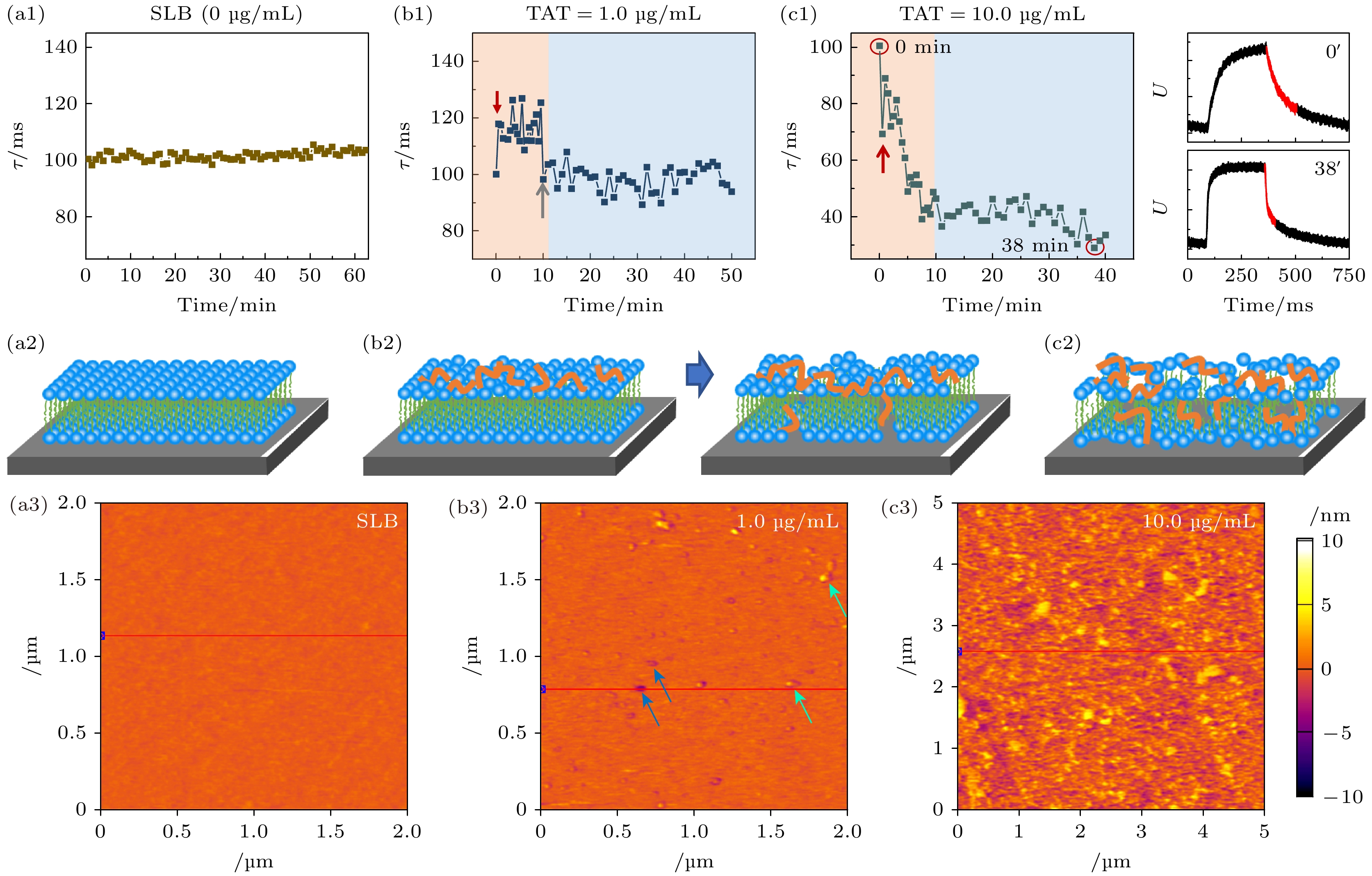
图 7 TAT多肽与磷脂膜相互作用过程的实时分析 (a1), (b1), (c1)不同浓度TAT (0, 1.0, 10.0 μg/mL)与DOPC膜相互作用过程的代表性τ-t曲线, 多肽-膜重构阶段以浅蓝色背景标记; (c1)右图为两个代表时刻(以红色圈标出)对应的电压脉冲; (a2), (b2), (c2) 对应的膜结构示意图, (b2)中蓝色和绿色箭头分别标出膜上的孔洞和吸附的颗粒; (a3), (b3), (c3) TAT孵育后DOPC 平板膜的AFM形貌图. 图片引用和修改自文献[44]
Figure 7. Real-time monitoring the interactions between TAT and a supported DOPC bilayer membrane using photo-voltage transient test. (a1), (b1), (c1) Representative τ-t profiles upon TAT exposure at 0, 1.0 and 10.0 μg/mL, respectively. The membrane reconstruction process is marked with a light blue background. The voltage pulses corresponding to the two circled points are representatively shown. The right figure shows the voltage pulses corresponding to two representative moments (marked in red circles). (a2), (b2), (c2) Corresponding membrane structures, blue and green arrows respectively mark the pores and adsorbed particles on the membrane in (b2). (a3), (b3), (c3) AFM topographies of DOPC SLBs after TAT incubation. Images are cited and modified from Ref. [44].
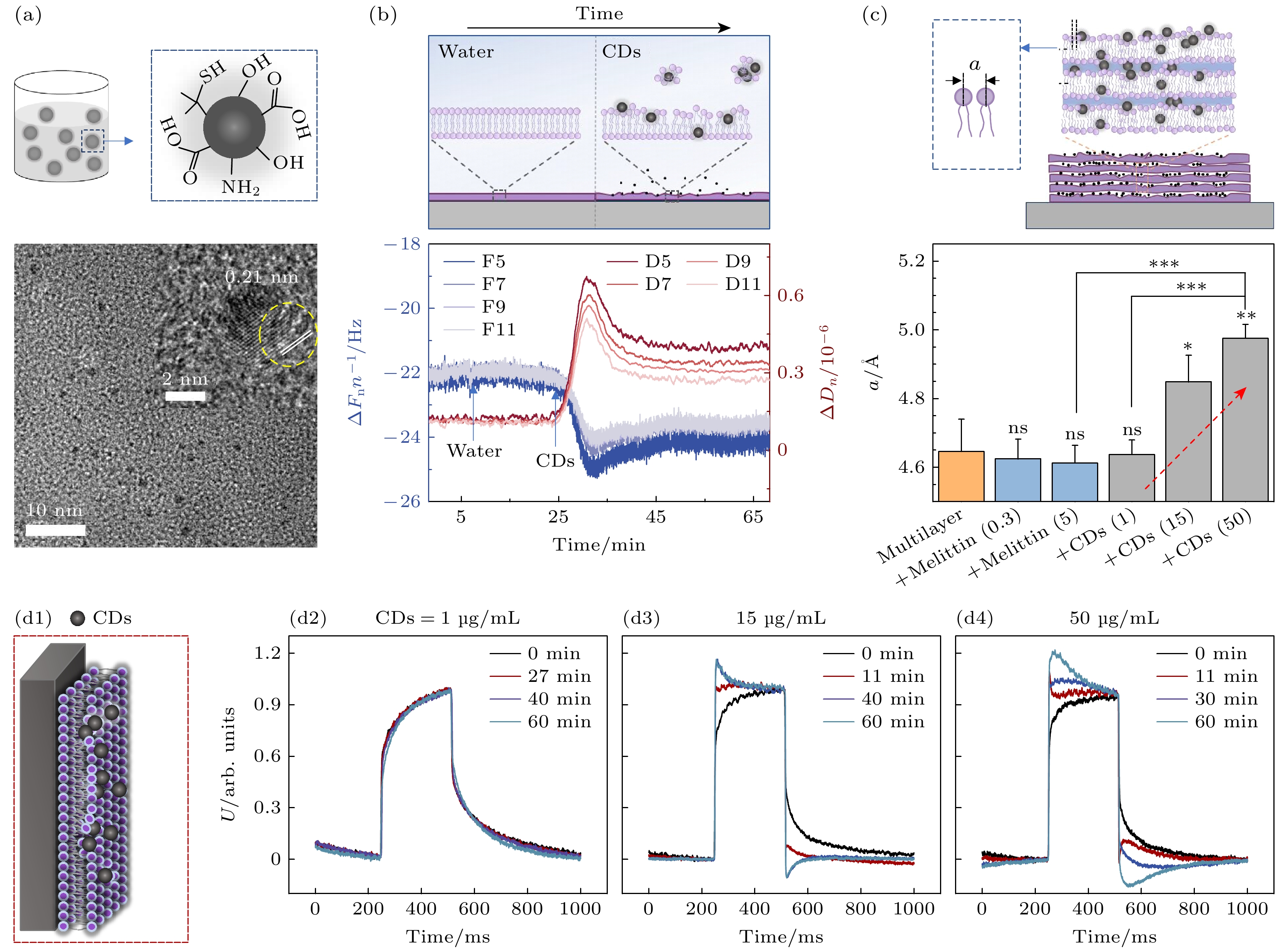
图 8 弱负电CDs对磷脂膜结构和电性质的扰动情况分析 (a)碳点的表面基团示意图及透射电子显微镜表征; (b)利用QCM-D实时记录CDs (浓度为50 μg/mL)与SLB的相互作用过程及CDs导致的脂膜双层结构不对称破坏, 箭头表示使用蠕动泵在10 mL/min的流速和25 ℃下向SLB中添加超纯水或CD的时间; (c)同步辐射XRD分析CDs对磷脂多层膜的层内磷脂间距(a)的影响, 原始溶液含有脂质(浓度为200 μg/mL), CDs浓度分别为1, 15, 50 μg/mL; (d)光电压瞬态实验分析CDs对SLB电性质的影响, 显示了典型的输出电压分布, 描绘了引入不同浓度CD后SLB的时间演变; (b)—(d)对应显示了可能的CDs导致膜结构扰动情况. 图片引用和修改自文献[45]
Figure 8. Impact of CDs on the structure and electrical properties of phospholipid membranes. (a) Schematic diagram, transmission electron microscopy (TEM) and high-resolution TEM images illustrating the morphology of CDs. (b) Asymmetric disruption of the two leaflets of bilayer membrane due to CD adsorption and perturbation under real-time QCM-D analysis, where arrows indicate the addition time of ultrapure water or CDs (at 50 μg/mL) to a SLB using a peristaltic pump at a flow rate of 10 mL/min and 25 ℃. (c) Synchrotron radiation XRD analysis of the CD-lipid composite structure. XRD analysis was performed on films composed of mixtures of DOPC lipids and varying concentrations of CDs or melittin, and the in-plane inter-lipid spacing, a, was demonstrated. The pristine solution contained lipids (at a concentration of 200 μg/mL), CDs ranging from 1 to 50 μg/mL. (d) The impact of CDs on the electrical properties of SLBs under photo-voltage analysis. Typical output voltage profiles were shown depicting the temporal evolution of the SLB subsequent to the introduction of CDs at different concentrations. (b)–(d) corresponding display of possible CDs causing membrane structure disturbance. Images are cited and modified from Ref. [45].
-
[1] Garcia-Lopez V, Chen F, Nilewski L G, Duret G, Aliyan A, Kolomeisky A B, Robinson J T, Wang G, Pal R, Tour J M 2017 Nature 548 567 doi: 10.1038/nature23657 [2] Dou Y J, Li J, Yuan B, Yang K 2014 Appl. Surf. Sci. 296 95 doi: 10.1016/j.apsusc.2014.01.050 [3] De Poli M, Zawodny W, Quinonero O, Lorch M, Webb S J, Clayden J 2016 Science 352 575 doi: 10.1126/science.aad8352 [4] Dawson K A, Salvati A, Lynch I 2009 Nat. Nanotechn. 4 84 doi: 10.1038/nnano.2008.426 [5] Roiter Y, Ornatska M, Rammohan A R, Balakrishnan J, Heine D R, Minko S 2008 Nano Lett. 8 941 doi: 10.1021/nl080080l [6] Alexeev A, Uspal W E, Balazs A C 2008 ACS Nano 2 1117 doi: 10.1021/nn8000998 [7] Michel J P, Wang Y X, Kiesel I, Gerelli Y, Rosilio V 2017 Langmuir 33 11028 doi: 10.1021/acs.langmuir.7b02864 [8] Xia H, Zhao Y 2020 Artif Cells Nanomed. Biotechnol. 48 197 doi: 10.1080/21691401.2019.1699826 [9] Shadmani P, Mehrafrooz B, Montazeri A, Naghdabadi R 2020 J. Phys. Condens. Matter 32 115101 doi: 10.1088/1361-648X/ab5a14 [10] Last N B, Miranker A D 2013 Proc. Natl. Acad. Sci. U S A 110 6382 doi: 10.1073/pnas.1219059110 [11] Weissmann G, Hirschhorn R, Krakauer K 1969 Biochem. Pharmacol. 18 1771 doi: 10.1016/0006-2952(69)90167-1 [12] Barbasz A, Ocwieja M, Roman M 2017 Colloid. Surf. B 156 397 doi: 10.1016/j.colsurfb.2017.05.027 [13] Vijayan V, Uthaman S, Park I K 2018 Polymers (Basel) 10 983 doi: 10.3390/polym10090983 [14] Han M L, Velkov T, Zhu Y, Roberts K D, Le Brun A P, Chow S H, Gutu A D, Moskowitz S M, Shen H H, Li J 2018 ACS Chem. Biol. 13 121 doi: 10.1021/acschembio.7b00836 [15] Khondker A, Dhaliwal A K, Saem S, Mahmood A, Fradin C, Moran-Mirabal J, Rheinstadter M C 2019 Commun. Biol. 2 67 doi: 10.1038/s42003-019-0297-6 [16] Ista L K, Yu Q, Parthasarathy A, Schanze K S, Lopez G P 2016 Biointerphases 11 019003 doi: 10.1116/1.4939239 [17] Xu Y C, Lin J X, Cui T, Srinivas S, Feng Y J 2018 J. Biol. Chem. 293 4350 doi: 10.1074/jbc.RA117.000924 [18] Keiderling T A, Lakhani A 2018 Chirality 30 238 doi: 10.1002/chir.22799 [19] Eun H J, Min A, Jeon C W, Yoo I, Heo J, Kim N J 2020 J. Phys. Chem. Lett. 11 4367 doi: 10.1021/acs.jpclett.0c01081 [20] Schwaighofer A, Alcaraz M R, Araman C, Goicoechea H, Lendl B 2016 Sci. Rep. 6 33556 doi: 10.1038/srep33556 [21] Hou S G, Liang L, Deng S H, Chen J F, Huang Q, Cheng Y, Fan C H 2014 Sci. China-Chem. 57 100 doi: 10.1007/s11426-013-5014-6 [22] Li Y, Qian Z Y, Ma L, Hu S X, Nong D G, Xu C H, Ye F F, Lu Y, Wei G H, Li M 2016 Nat. Commun. 7 12906 doi: 10.1038/ncomms12906 [23] Ma D, Hou W, Yang C, Hu S, Han W, Lu Y 2021 Biophys. Rep. 7 490 doi: 10.52601/bpr.2021.210030 [24] Xu C, Ma W D, Wang K, He K J, Chen Z L, Liu J J, Yang K, Yuan B 2020 J. Phys. Chem. Lett. 11 4834 doi: 10.1021/acs.jpclett.0c01169 [25] Wu J F, Xu C, Ye Z F, Chen H B, Wang Y P, Yang K, Yuan B 2023 Small 19 2301713 doi: 10.1002/smll.202301713 [26] Inoue O, Hasumi K 2019 J. Micro/Nanolith. MEM. 18 021206 doi: 10.1117/1.JMM.18.2.021206 [27] Zhang B, Ma X L 2019 J. Mater. Sci. Technol. 35 1455 doi: 10.1016/j.jmst.2019.01.013 [28] Eswara S, Pshenova A, Yedra L, Hoang Q H, Lovric J, Philipp P, Wirtz T 2019 Appl. Phys. Rev. 6 021312 doi: 10.1063/1.5064768 [29] Kosari A, Zandbergen H, Tichelaar F, Visser P, Terryn H, Mol A 2019 Corrosion 76 4 doi: 10.5006/3369 [30] Li S Q, Chang Y J, Wang Y M, Xu Q, Ge B H 2020 Micron 130 102813 doi: 10.1016/j.micron.2019.102813 [31] Chen P Y, Huang Y, Zhai X B, Huang Z H, Ma G H, Wei W, Yan L T 2019 Sci. Adv. 5 aaw3192 doi: 10.1126/sciadv.aaw3192 [32] Xiao S F, Lu X M, Gou L, Li J L, Ma Y Q, Liu J J, Yang K, Yuan B 2019 Carbon 149 248 doi: 10.1016/j.carbon.2019.04.067 [33] Suk H J, Boyden E S, van Welie I 2019 J. Neurosci. Methods 326 108357 doi: 10.1016/j.jneumeth.2019.108357 [34] de Pauli M, Gomes A M C, Cavalcante R L, Serpa R B, Reis C P S, Reis F T, Sartorelli M L 2019 Electrochim. Acta 320 134366 doi: 10.1016/j.electacta.2019.06.059 [35] Marchioro A, Bischoff M, Lutgebaucks C, Biriukov D, Predota M, Roke S 2019 J. Phys. Chem. C 123 20393 doi: 10.1021/acs.jpcc.9b05482 [36] Rao Y, Song D H, Turro N J, Eisenthal K B 2008 J. Phys. Chem. B 112 13572 doi: 10.1021/jp802499w [37] Eisenthal K B 2006 Chem. Rev. 106 1462 doi: 10.1021/cr0403685 [38] Dou Y J, Li W W, Xia Y, Chen Z L, Wu Z L, Ge Y K, Lin Z, Zhang M L, Yang K, Yuan B, Kang Z H 2020 ACS Appl. Bio Mater. 4 620 doi: 10.1021/acsabm.0c01180 [39] Xia Y, Sun S Q, Zhang Z H, Ma W D, Dou Y J, Bao M M, Yang K, Yuan B, Kang Z H 2022 Phys. Chem. Chem. Phys. 24 5360 doi: 10.1039/D1CP05598D [40] Ge Y K, Liu J J, Dou Y J, Chen Z L, Li Z H, Yang K, Yuan B, Kang Z H 2022 Colloid. Surf. B 209 112161 doi: 10.1016/j.colsurfb.2021.112161 [41] Yuan B, Liu J J, Deng Z X, Wei L, Li W W, Dou Y J, Chen Z L, Zhang C, Xia Y, Wang J, Zhang M L, Yang K, Ma Y Q, Kang Z H 2021 NPG Asia Mater. 13 18 doi: 10.1038/s41427-021-00287-y [42] Ma W, Sun S Q, Li W W, Zhang Z H, Lin Z, Xia Y, Yuan B, Yang K 2020 Langmuir 36 7190 doi: 10.1021/acs.langmuir.0c00194 [43] Wei L, Tu W Q, Xu Y, Xu C, Dou Y J, Ge Y K, Sun S Q, Wei Y S, Yang K, Yuan B 2024 ACS Nano 18 18650 doi: 10.1021/acsnano.4c05265 [44] Sun S Q, Xia Y, Liu J, Dou Y J, Yang K, Yuan B, Kang Z 2022 J. Colloid Interface Sci. 609 707 doi: 10.1016/j.jcis.2021.11.076 [45] Li W W, Ma Y, Ou L P, Xu C, Wei Y S, Yang K, Yuan B 2024 J. Hazard. Mater. 465 133382 doi: 10.1016/j.jhazmat.2023.133382 [46] Wei Y S, Chen H B, Li Y X, He K J, Yang K, Pang H B 2022 ACS Nano 16 5885 doi: 10.1021/acsnano.1c11068 [47] Ou L P, Chen H B, Yuan B, Yang K 2022 ACS Nano 16 18090 doi: 10.1021/acsnano.2c04774 -
计量
- 文章访问数: 1112
- HTML全文浏览数: 1112
- PDF下载数: 10
- 施引文献: 0