
 首页
首页 登录
登录 注册
注册



-
能源作为社会发展的重要驱动力, 始终受到各国的高度重视. 经济的持续增长以及人民生活水平的不断提高, 都依赖于充足的能源供应. 传统的化石燃料, 如煤、石油、天然气等储量有限且短期内不可再生, 同时其燃烧产生的二氧化碳等物质加剧了全球变暖和环境污染[1–3]. 因此, 寻求可持续的绿色能源, 如氢能、太阳能、风能、地热能和海洋能等迫在眉睫. 氢作为能源载体的优势在于其在自然界的储藏量丰富且分布广泛, 氢的能量密度及燃烧热值较传统化石燃料更高, 而且氢燃烧的产物是水, 从而有效减少了环境污染[4,5]. 储氢技术是氢能应用的关键环节, 目前常见的储氢方式包括高压气态储氢和低温液态储氢等, 它们在储氢密度、安全性、成本等方面存在诸多挑战, 难以满足现代工业化和大规模应用的需求. 因此开发新型高效的固体储氢材料成为当前能源领域的热点之一[6–8].
硼基纳米材料具有大比表面积、高孔隙率和轻质等特性, 在固体储氢领域得到了广泛的研究[9,10], 特别是硼纳米团簇由于具有独特的电子结构和几何构型, 近年来成为研究者重点关注的对象之一. 研究发现, 通过金属原子修饰能够显著提高纳米材料的储氢性能[11]. 例如, 金属功能化的硼富勒烯[12]、硼氮纳米片[13–15]以及硼纳米管[16,17]等在室温下表现出良好的储氢性能. 碱土金属修饰的B38富勒烯能够形成稳定结构, 其中在Ca修饰的B38富勒烯中, 每个Ca原子能吸附5个氢分子, 平均吸附能为0.24 eV/H2, 储氢密度达到6.47% (质量分数) [18]. 在过渡金属Sc原子修饰的B40富勒烯中, Sc原子稳定地结合在B40的空腔外, 每个Sc原子能吸附5个氢分子, 平均吸附能处在0.33—0.58 eV/H2范围[19]. 研究表明, 在室温条件下金属修饰的Bn (n = 3—10)团簇比中等尺寸或者大尺寸硼纳米结构表现出更优异的储氢性能. 例如, 氢以分子形式吸附在Li原子修饰的B14团簇周围, 平均吸附能处在0.12—0.14 eV/H2范围内, 其中Li5B14在室温下表现出最大的氢储存容量(质量分数13.89%)[20]. 对于碱土金属原子(Be, Mg, Ca)修饰的B7团簇, 每个金属原子可吸附5—7个氢分子, 吸附能为0.10—0.48 eV/H2, 最高储氢密度可达到23.03%(质量分数)[21]. 与碱金属和碱土金属相比, 过渡金属原子质量较大, 在其修饰的Bn (n = 3—10)团簇中, 吸附等量氢分子时, 氢分子质量占比较小. 例如, M2B7(M = Fe, Co, Ni)团簇能够吸附12个氢分子, 储氢密度为11.11%—11.43%(质量分数)[22]. 值得注意的是, 碱金属Na原子因具有轻质特性和适中的电负性, 其修饰的硼团簇也展现出良好的储氢性能. 研究发现, Na原子能够稳定结合在B28团簇的空心位点, 结合能远大于其块体内聚能, 有效避免了金属原子的聚集现象. Na修饰的B28团簇可吸附多达48个氢分子, 平均吸附能在0.2—0.6 eV/H2范围内, 储氢重量密度为7.99%(质量分数)[23]. Ruan等[24]研究了NanBm (m + n = 6)团簇的最稳定异构体的储氢性能, 发现Na3B3具有最高的储氢能力. 此外, Na原子修饰的二维硼单层中, Na原子稳定地结合在六元环空位两边, 每个Na原子能够有效吸附7个氢分子, 吸附能为0.148 eV/H2, 实现了8.28%(质量分数)的高储氢量[25]. 由此可见, Na原子修饰的硼纳米结构具有很好的储氢性能, 为开发新型轻金属修饰储氢材料提供了重要的理论依据.
本文利用密度泛函理论方法对碱金属Na原子修饰的(BnNa2, n = 3—10)团簇的储氢性能进行研究, 并通过分子动力学模拟研究了这些复合团簇在不同温度下的脱氢规律. Na原子修饰的Bn (n = 3—10)团簇不仅具有良好的储氢性能, 而且在常温条件下大部分团簇能够实现极高的脱氢效率, 表明Na原子修饰的Bn (n = 3—10)团簇可以作为一种潜在的候选储氢材料.
-
本文的理论计算采用三阶混合密度泛函B3LYP方法, 其中交换泛函包含20%的Hartree-Fock精确交换、8%的Slater交换和72%的Becke梯度校正交换, 相关泛函采用Lee-Yang-Parr泛函. 针对钠掺杂硼团簇体系的特点, 选择6-311+G(d, p)分裂价基组进行几何结构优化与能量计算, 其包含弥散函数和极化函数的特点可有效描述分子轨道的空间分布及弱相互作用体系[26–28]. 为准确表征体系中可能存在的范德瓦耳斯相互作用, 在所有计算中引入Grimme提出的D3(BJ)色散校正方法, 该方法通过Becke-Johnson阻尼函数有效降低远程相互作用的高估问题[29]. 几何优化过程中采用Berny算法进行势能面搜索, 收敛标准设定为能量变化小于1×10–5 Hartree, 最大力小于4.5×10–4 Hartree/Bohr, 最大位移小于1.8×10–3 Å. 通过振动频率分析确认所有优化构型均处于势能面极小点(无虚频), 其中频率计算采用解析二阶导数法. 基于优化后的基态构型, 利用Multiwfn软件包(3.8版)进行态密度(DOS)分析[30]. 通过中心原子密度矩阵传播方法(ADMP)进行分子动力学模拟研究Na掺杂Bn (n = 3—10)团簇表面吸附氢分子的热稳定性和可逆性, 模拟时间步长设为1 fs, 总时长为1000 fs. 所有计算通过Gaussian16程序包完成[31].
Na原子在Bn团簇上的平均结合能为:
式中,
$ E({\text{Na}}) $ ,$ E({{\text{B}}_n}) $ 和$ E({{\text{B}}_n}{\text{N}}{{\text{a}}_2}) $ 分别为单个Na原子的能量、Bn团簇和BnNa2团簇的总能量.氢分子在BnNa2团簇上的平均吸附能:
式中,
$ E({{\text{H}}_{2}}) $ 表示单个氢分子的能量,$ E({{\text{B}}_n}{\text{N}}{{\text{a}}_{2}}) $ 和$ E({{\text{B}}_n}{\text{N}}{{\text{a}}_{2}}{{(}{{\text{H}}_{2}}{)}_m}) $ 分别表示BnNa2团簇的总能量和BnNa2团簇吸附$m$ 个氢分子后的总能量.其中,
$ M({{\text{H}}_{2}}) $ 与$ M({{\text{B}}_n}{\text{N}}{{\text{a}}_{2}}) $ 分别表示单个氢分子的质量与BnNa2团簇的质量. -
首先, 对Bn (n = 3—10)团簇的结构进行优化, 计算得到的最低能结构与前期文献[32]报道的结果一致, 其中硼团簇的每个原子平均结合能和平均B—B键长分别处于2.854—4.632 eV和1.521—1.605 Å之间, 与文献[32]报道的相应结果最大偏差分别为3.9%和7.3%. 接下来, 在纯硼团簇表面的不同初始结合位上放置Na原子进行结构优化, 确定了Na原子的最佳结合位点, 获得了BnNa2 (n = 3—10)的稳定结构. 如图1所示, 在B3Na2团簇中, 一个Na原子位于三元环面位上方, 另一个Na原子位于其中一个桥位侧下方; 在B4Na2和B5Na2团簇中, 两个Na原子分别结合在B四元环和五元环的顶点原子的两侧, 且与B团簇处在同一平面内; 在B6Na2和B7Na2团簇中, 两个Na原子都位于B团簇准平面两侧, 其中一侧的Na原子位于环中心上方, 另一侧的Na原子分别位于顶点B原子的顶位和边缘桥位上; 在B8Na2和B9Na2团簇中, 两个Na原子都位于B团簇平面两侧, 其中B8Na2团簇的两个Na原子位于环面中心B原子顶位, B9Na2团簇的Na原子偏向环面的空心位; 在B10Na2团簇中, 两个Na原子分别结合在环边缘的两个B原子顶位上.
表1给出了Na原子修饰的Bn (n = 3—10)团簇的相关参数. 在Na原子修饰的Bn (n = 3—10)结构中, Na原子与B原子之间平均距离在2.355—2.669 Å之间, 比B14Lin (n = 1—5)团簇中Li原子到B原子的平均距离(2.098—2.188 Å)略微增大[20], 这是由于Na原子的半径比Li原子的大. 两个Na原子之间的平均距离介于4.043—6.574 Å之间, Na原子之间的距离较远, 且在主体团簇上的平均结合能介于1.876—2.967 eV之间, 明显大于其块体的内聚能(1.113 eV), 有效避免了Na原子在团簇表面可能出现聚集行为[33], 为氢分子的吸附提供了充足的空间和吸附位点. 另外, 从表1可以看出, BnNa2 (n = 3—10)团簇的HOMO-LUMO能隙处于1.867—3.546 eV之间.
NBO电荷分析表明, 在BnNa2 (n = 3—10)团簇中, Na原子带有正电荷0.837e—0.979e, 这与单个Na原子修饰Bn (n = 3—10)团簇中Na原子电荷量(0.79e —0.98e)基本一致[34], 说明当Na原子结合到B团簇上时, Na原子的部分电子转移到B原子上, 使得Na原子带正电荷, 而B原子带负电荷. 例如, 在B3Na2结构中, Na原子上的电荷为0.837e, 而B原子上的电荷为–0.558e. 为了进一步分析Na原子与B团簇之间的作用机理, 以B3Na2团簇为例, 计算了其电子态密度, 如图2所示. 从图2可以看出, Na的3s3p轨道与B的2s2p轨道在能级–7.436, –5.649和–4.768 eV处发生了微小重叠, 说明在Na原子与B团簇之间形成的局域电场作用下, Na的3p轨道发生了分裂, 与B的2p轨道重叠使得少量的电子回赠到Na的3p轨道上, Na的3p轨道参与了弱的成键, 这样Na原子与B团簇结合形成了以离子键为主和共价作用为辅的成键特征.
-
首先, 在BnNa2 (n = 3—10)团簇中的每个Na原子周围放置一个氢分子, 氢分子的H—H键长设置为自由氢分子的键长0.743 Å, 经过优化之后氢分子能够以侧位方式吸附在Na原子上, 氢分子与Na原子之间的平均距离介于2.379—2.492 Å之间, 氢分子的平均吸附能处在0.063—0.127 eV范围内, 氢分子的H—H键长被略微拉长(0.748—0.750 Å), 这与B14Lin (n = 1—5)团簇吸附的氢分子H—H键长范围完全一致[20]. 接着, 按照以上方式继续在每个主体团簇的两个Na原子周围逐个添加氢分子, 依次进行结构优化, 探究这些主体团簇对氢分子的吸附规律和储氢性能. 计算结果表明, 当每个团簇周围放置12个氢分子后, 进行结构优化发现第11和12个氢分子已经远离Na原子, 说明Na原子吸附氢分子已经达到饱和. 因此, 在BnNa2 (n = 3—10)团簇中, 每个结构最多能吸附10个氢分子, 即每个Na原子周围吸附5个氢分子, 储氢密度可达到11.57%—20.45%(质量分数), 这远大于碱土金属Mg原子和过渡金属Sc原子修饰硼团簇的储氢密度, BnMg2 (n = 4—10)最多能够吸附2—4个氢分子, 储氢密度为2.48%—8.10%(质量分数)[9], BnSc2 (n = 3—10)团簇能够饱和吸附2—8个氢分子, 储氢密度为3.19%—9.43%(质量分数)[35]. BnNa2 (n = 3—10)团簇吸附氢分子的几何结构如图3所示, 吸氢结构的相关参数已列在表2中. 氢分子的平均吸附能随吸附氢分子数量的变化如图4所示. 在这些结构中, 随着吸附氢分子数量的增大, 氢分子的吸附能逐渐减小, 氢分子到Na原子的距离有所增大, 这是因为氢分子数量越大, 它们之间的排斥作用越强, 使得部分氢分子与Na原子距离较远, 吸附能逐渐减小. B3Na2结构对氢分子的吸附作用最小, 平均吸附能处于0.061—0.063 eV/H2之间, 而B10Na2结构对氢分子的吸附作用最大, 平均吸附能处于0.095—0.111 eV/H2之间, 当BnNa2 (n = 3—10)团簇饱和吸附氢分子时, 氢分子的平均吸附能处于0.063—0.095 eV/H2范围内, 氢分子到Na原子的平均距离处在2.535—2.590 Å范围. 表3给出了BnNa2 (n = 3—10)团簇吸附氢分子前后的NBO电荷分布情况. 当Na原子与Bn (n = 3—10)团簇结合时, Na原子向B原子转移部分电子, 使Na原子带正电荷, B原子带负电荷. 当氢分子接近主体团簇时, 在其周围形成的局域电场中被适度极化, 且带有极少量的电荷, 通过静电相互作用吸附在Na原子周围. 因此, BnNa2 (n = 3—10)团簇吸附的所有氢分子的H—H键长都被稍微拉长(0.748—0.750 Å), 但是没有发生断裂, 氢仍然以分子形式吸附. 值得注意的是, BnNa2 (n = 3—10)吸附氢分子之后, Na原子到B原子的平均距离处于2.379—2.672 Å范围, 比吸附氢分子之前的Na–B距离略有增大, 说明氢分子的吸附对BnNa2 (n = 3—10)团簇的几何结构影响很小.
最后, 通过中心原子密度矩阵传播方法对BnNa2(H2)10 (n = 3—10)体系进行分子动力学模拟, 进一步探究了这些结构在不同温度下的脱氢效率. 模拟温度为77, 200和300 K, 时间步长为1 fs, 总时长为1000 fs. 如图5所示, 在77, 200和300 K温度下, B3Na2(H2)10体系的第1个氢分子脱附时间分别为78, 41和32 fs.这说明温度越高, 第1个氢分子脱离主体团簇的时间越短. 在77 K温度下, 时间为261—1000 fs内, B3Na2团簇仍然吸附3个氢分子, 储氢密度为7.16% (质量分数), 在200 K温度下, 时间为78—1000 fs, 只有1个氢分子吸附在主体团簇周围, 储氢密度降低至2.51%(质量分数). 当温度升高至300 K, 时间为80 fs时, 所有氢分子均已从B3Na2团簇周围脱离, 这说明在有限时间里, 温度越高, 氢分子的脱附量越大, 在常温条件下, 所有氢分子都能够在更短时间内完全脱附, 其他团簇的脱氢过程也有类似的规律.
接下来, 重点探讨在常温条件下(300 K)BnNa2(H2)10 (n = 3—10)团簇的脱氢速率和氢分子的脱附率. 图6展示了300 K温度下BnNa2(H2)10 (n = 3—10)体系的第1个和最后1个氢分子脱离主体团簇的时间. 从图6可以看出, B3Na2(H2)10体系脱附第1个氢分子的时间最短(32 fs)且10个氢分子全部脱附的时间也最短(80 fs), 说明B3Na2(H2)10团簇的脱氢速率最大; B4Na2(H2)10脱附第1个氢分子的时间最长(97 fs), 其他体系脱附第1个氢分子的时间比较集中(41—50 fs); B7Na2(H2)10团簇完全脱附氢分子的时间最长(262 fs), 脱附速率最小. 在模拟结束时(1000 fs), 除了B9Na2和B10Na2团簇各吸附1个氢分子之外(脱氢率为90%), 其他主体团簇吸附的所有氢分子都能够在更短时间内完全脱附, 实现了氢分子100%的脱附率. 正如前面所述, 氢分子主要通过静电相互作用吸附在主体团簇周围, 在所有饱和吸氢体系中, B3Na2(H2)10体系能够在最短时间内完全脱附, 主要是因为B3Na2团簇中Na原子和B原子的电荷量最小, 而且主体团簇尺寸较小, 其周围吸附位点距离更近, 氢分子之间的排斥作用较强, 因此这些氢分子容易脱附. 随着主体团簇尺寸增大, 氢分子间的排斥作用减小, 其吸附能逐渐增大, 氢分子的脱附速率减小. 图7给出不同温度下BnNa2(H2)10 (n = 3—10)体系的势能随时间的变化关系. 当氢分子脱附时, 它们与主体团簇之间的相互作用减弱, 导致体系势能降低, 由于脱附过程需要克服一定的能垒, 所以体系势能呈阶梯式下降, 每个台阶对应一个或一组氢分子脱附. 当所有氢分子脱附后, 体系势能趋于相对稳定, 但由于主体团簇自身的热振动, 仍会有较小的波动; 同时, 对于所有体系, 温度较低时, 势能波动较小, 温度越高, 体系的势能越大且势能变化幅度越大, 波动频率也增大.
-
本文采用密度泛函理论方法研究了Na原子修饰的Bn (n = 3—10)团簇的储氢性能. 结果表明, 当Na原子与Bn (n = 3—10)团簇结合时, Na原子向B原子转移电子, 使得Na原子带正电荷, 从而导致氢分子发生极化现象, 并通过静电相互作用吸附在Na原子周围.Na修饰的Bn (n = 3—10)团簇最多可以吸附10个氢分子, 平均吸附能介于0.063—0.095 eV/H2之间, 最大储氢密度可达到11.57%—20.45%(质量分数). 分子动力学模拟表明, 温度越高, 氢分子的脱附速率越大, 脱附量也越大, 在常温条件下, 时间为1000 fs内, 除了B9Na2和B10Na2团簇各吸附1个氢分子之外(脱氢率为90%), 其他主体团簇都实现了氢分子100%的脱附率. 可见, Na原子修饰的Bn (n = 3—10)团簇不仅有良好的储氢性能, 而且在常温条件下能够实现高效脱氢, 可以作为一种潜在的储氢候选材料.
Na原子修饰的Bn (n = 3—10)团簇的储氢性能
Hydrogen storage properties of Na-decorated Bn(n = 3–10) clusters
-
摘要: 采用密度泛函理论方法研究了Na原子修饰的Bn (n = 3—10)团簇的储氢性能. 结果表明, 两个Na原子能够与Bn团簇稳定地结合形成BnNa2 (n = 3—10)复合体. Na原子修饰的Bn团簇最多可以吸附10个氢分子, 平均吸附能处在0.063—0.095 eV/H2范围内, 最大储氢密度介于11.57%—20.45% (质量分数)之间. 分子动力学模拟表明, 温度越高, 氢分子的脱附速率越大, 脱附量也越大, 在常温条件下, BnNa2 (n = 3—8)团簇能够在短时间内(短于262 fs)实现完全脱氢, 因此, Na原子修饰的Bn团簇是一类极具潜力的储氢材料.Abstract: Hydrogen is widely regarded as an ideal alternative energy source because of its high efficiency, abundance, pollution-free and renewable properties. One of the main challenges is to find efficient materials that can store hydrogen safely with fast kinetics, favorable thermodynamics, and high hydrogen density under ambient conditions. The nanomaterial is one of the most promising hydrogen storage materials because of its high surface-to-volume rate, unique electronic structure and novel chemical and physical properties. In this study, the hydrogen storage properties of Na-decorated Bn (n = 3–10) clusters are investigated using dispersion-corrected density functional theory and atomic density matrix propagation (ADMP) simulations. The results show that Na atoms can stably bind to Bn clusters, forming BnNa2 complexes. The average binding energies (1.876–2.967 eV) of Na atoms on the host clusters are significantly higher than the cohesive energy of bulk Na (1.113 eV), which effectively prevents Na atoms from gathering on the cluster surface. Furthermore, when Na atoms bind to Bn (n = 3–10) clusters, electrons transfer from Na to B atoms, resulting in positively charged Na atoms. Hydrogen molecules are moderately polarized under the electric field and adsorbed around Na atoms through electrostatic interactions. The H–H bonds are slightly stretched but not broken. The Na-decorated Bn clusters can adsorb up to 10 hydrogen molecules with average adsorption energies of 0.063–0.095 eV/H2 and maximum hydrogen storage densities reaching 11.57%–20.45%. Almost no structural change is observed in the host clusters after adsorbing hydrogen. Molecular dynamics simulations reveal that the desorption rate of hydrogen molecules increases with temperature rising. At ambient temperature (300 K), BnNa2 (n = 3–8) clusters achieve complete dehydrogenation within 262 fs, while B9Na2 and B10Na2 clusters exhibit a dehydrogenation rate of 90% within 1000 fs. The Na-decorated Bn (n = 3–10) clusters not only exhibit excellent properties for hydrogen storage but also enable efficient dehydrogenation at ambient temperature. Thus, BnNa2 (n = 3–10) clusters can be regarded as highly promising candidates for hydrogen storage.
-
Key words:
- boron clusters /
- hydrogen storage performance /
- adsorption energy /
- density functional theory .
-

-
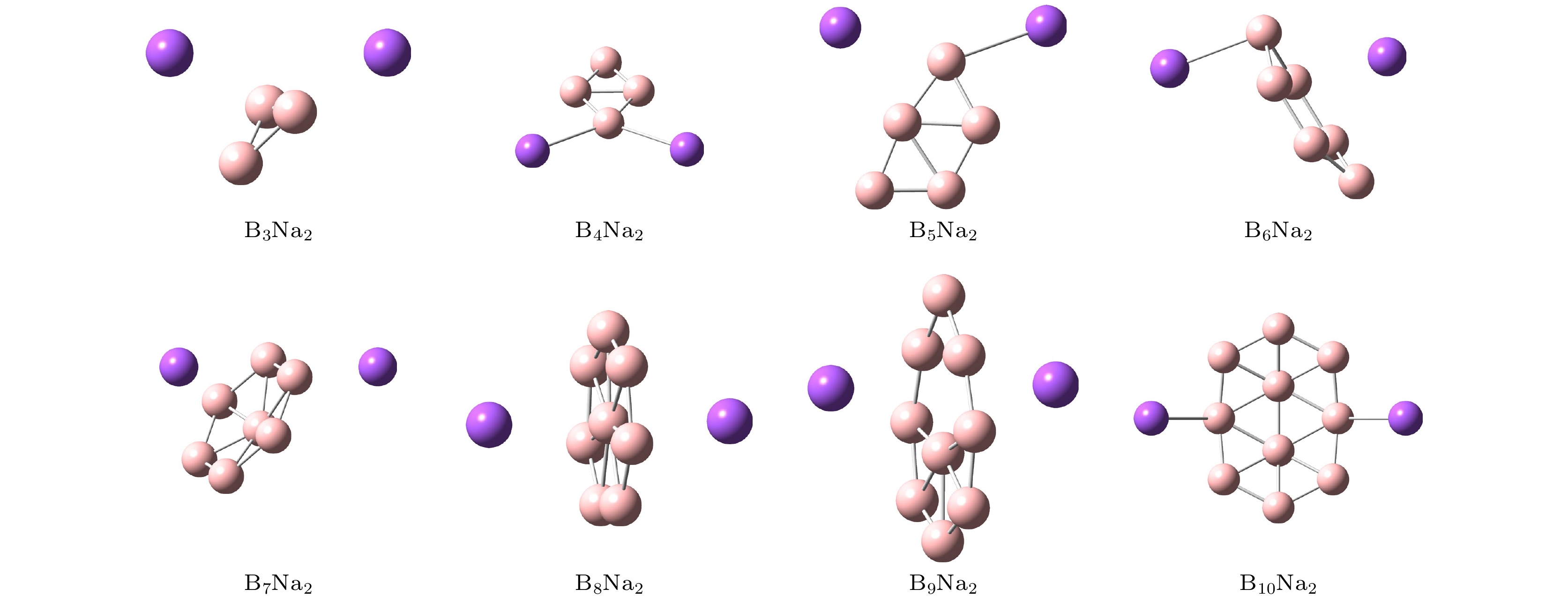
图 1 BnNa2 (n = 3—10)团簇的优化结构
Figure 1. Optimization structures of BnNa2 (n = 3–10) cluster.
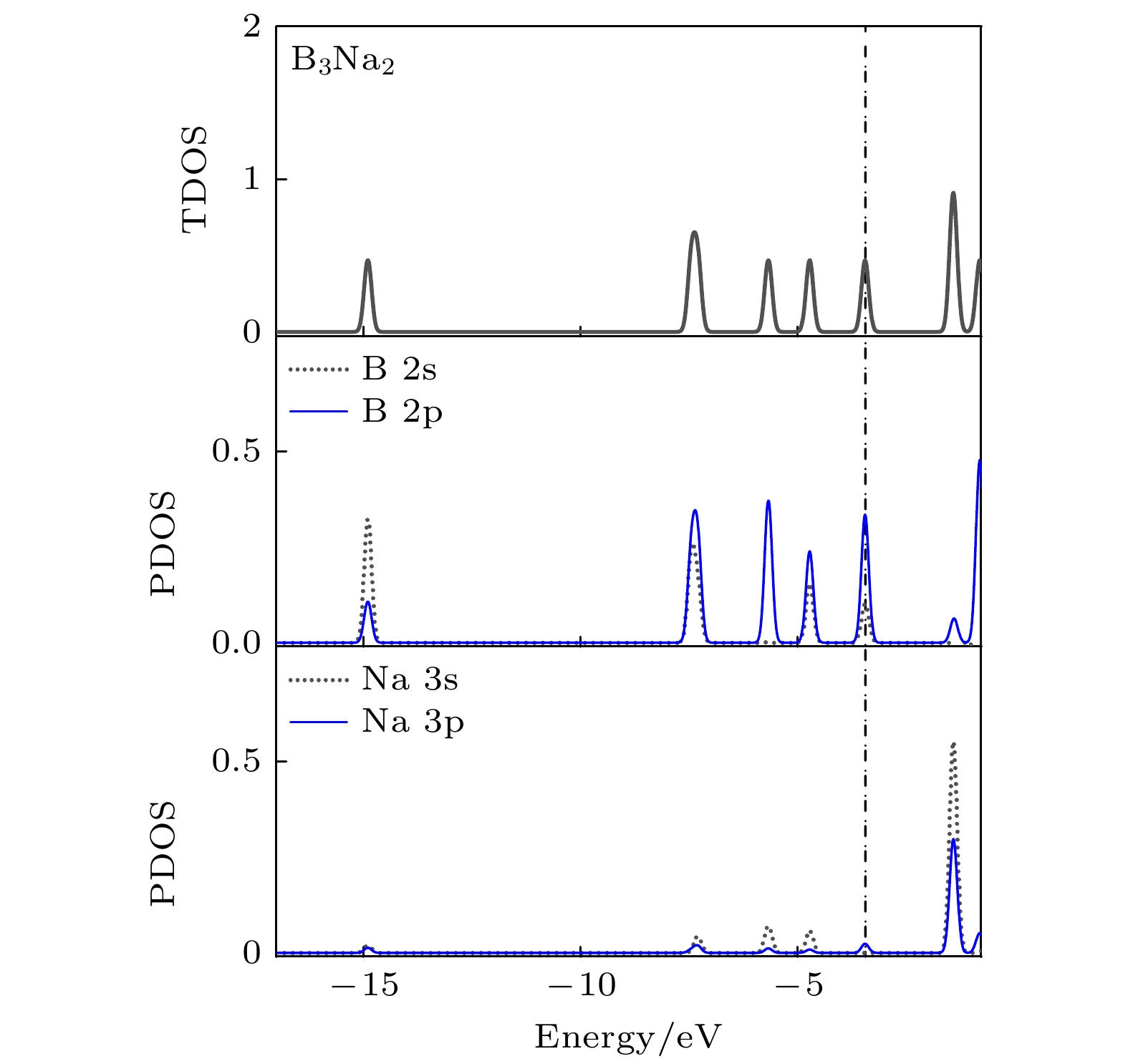
图 2 B3Na2的总态密度与分态密度, 其中垂直虚线表示HOMO能级的位置
Figure 2. TDOS and PDOS of B3Na2, the vertical dashed line indicates the HOMO level.
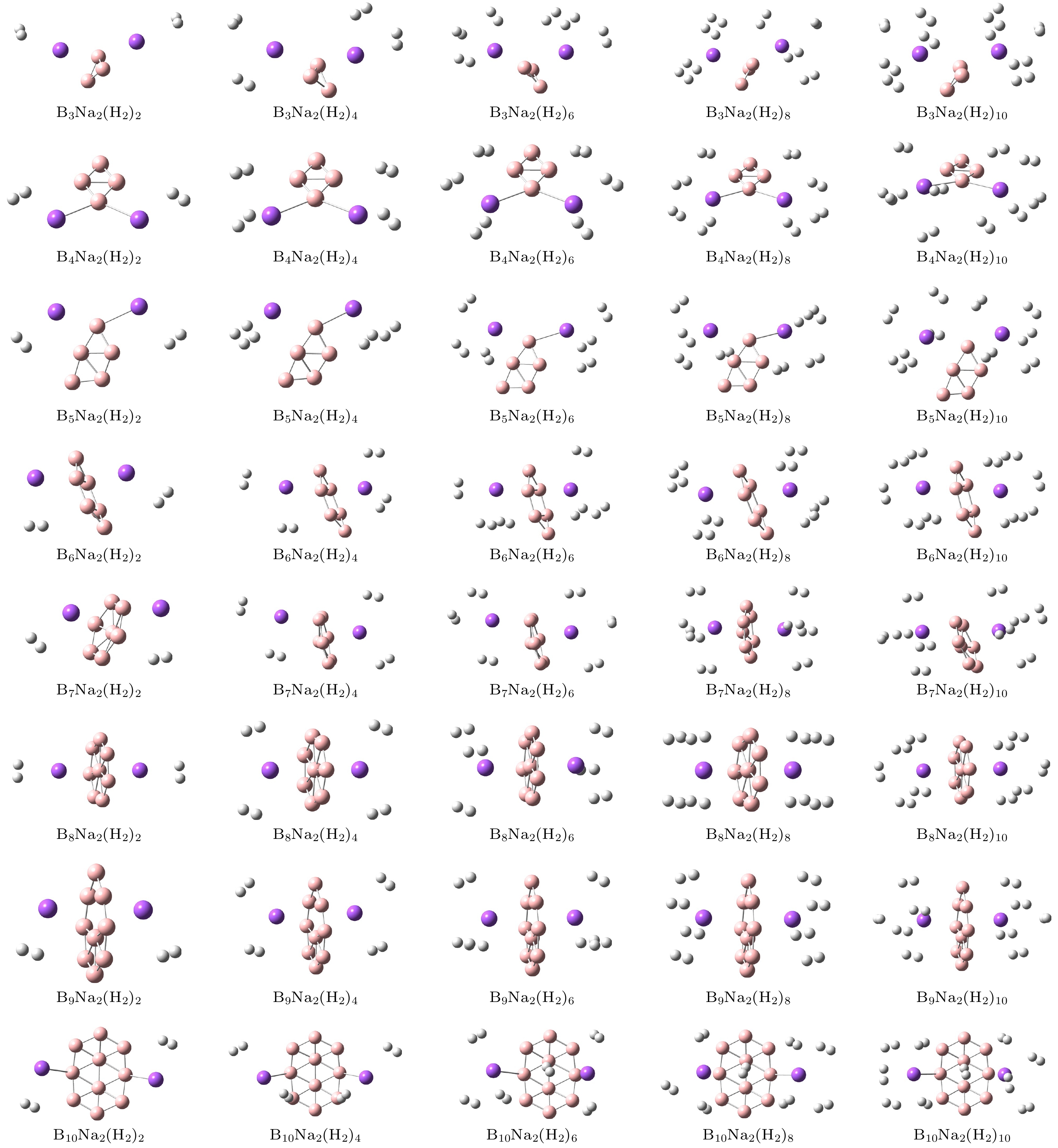
图 3 BnNa2 (n = 3—10)团簇的吸氢结构
Figure 3. Adsorption configurations of hydrogen molecules on BnNa2 (n = 3–10) clusters.
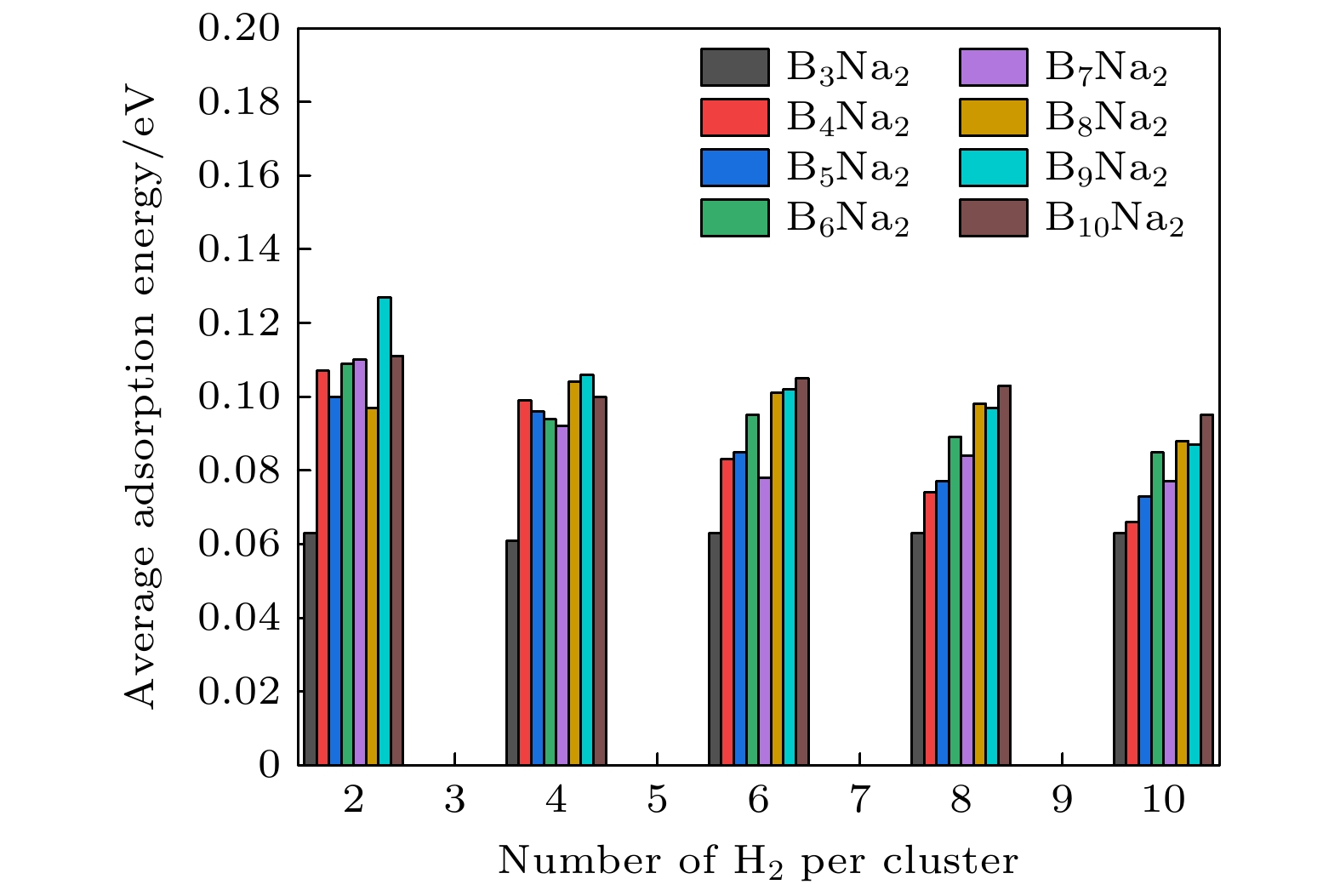
图 4 氢分子的平均吸附能随吸附氢分子数量的变化
Figure 4. The variation of the average adsorption energy with the number of adsorbed H2 molecules.
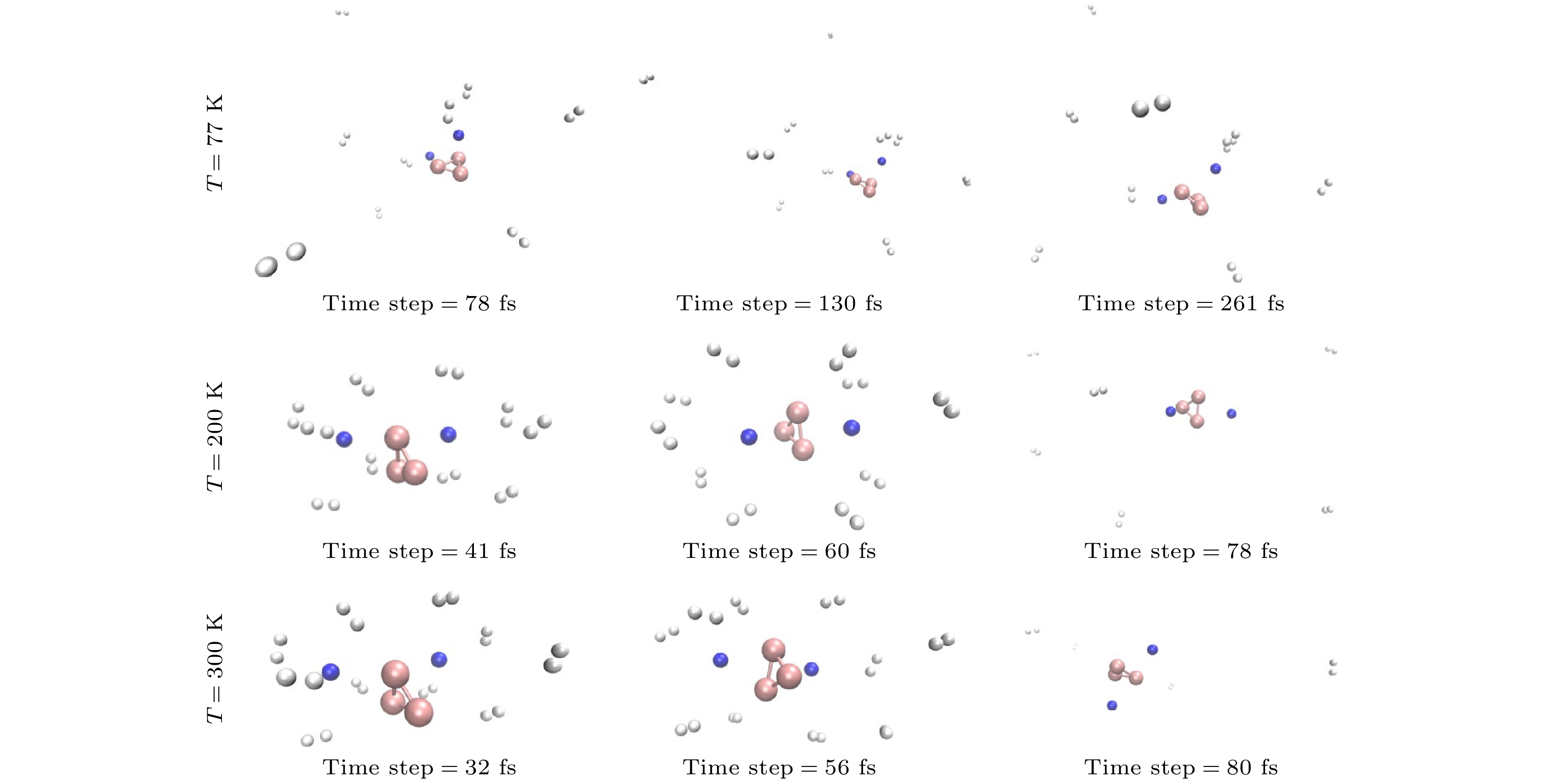
图 5 在77, 200和300 K温度下B3Na2(H2)10团簇的脱附状态
Figure 5. The desorption states of the B3Na2(H2)10 cluster at 77, 200 and 300 K.
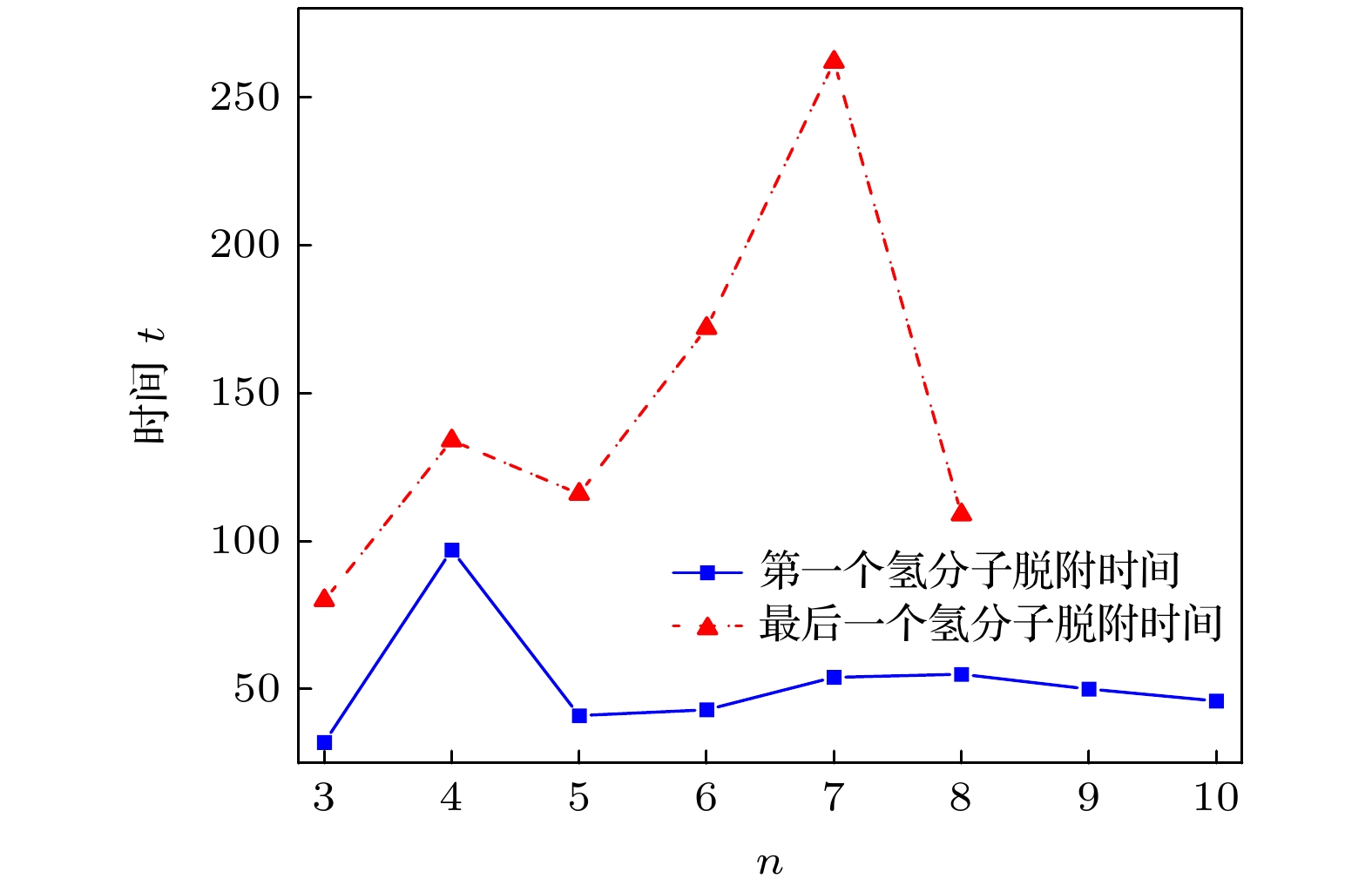
图 6 在300 K下, 第1个和最后1个氢分子脱离BnNa2(H2)10 (n = 3—10)团簇的时间
Figure 6. Desorption times of the first and last H2 molecules from BnNa2(H2)10 (n = 3–10) clusters at 300 K.
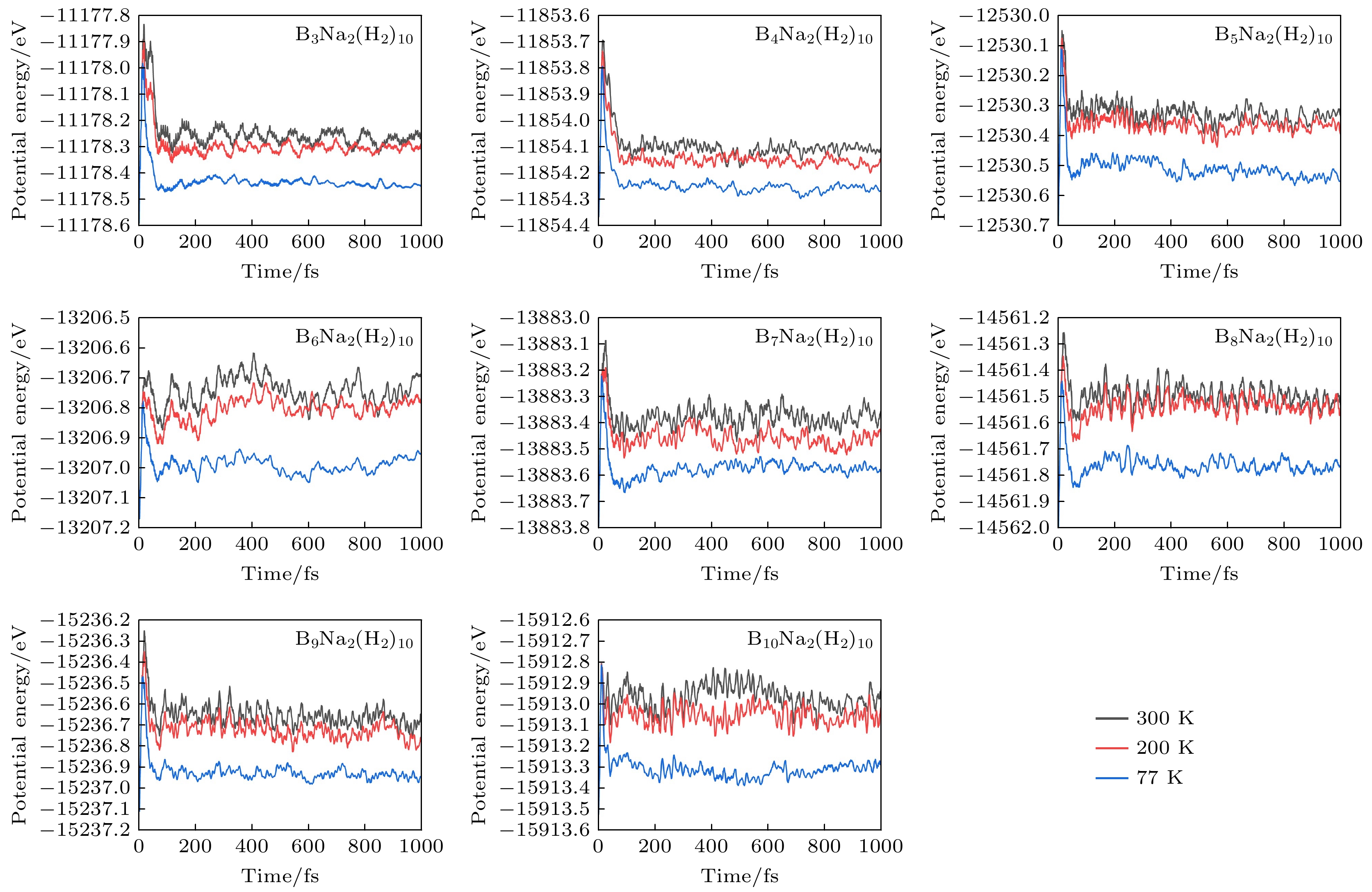
图 7 在77, 200和300 K下, BnNa2(H2)10体系的势能随时间的变化关系
Figure 7. The potential energy versus time for the BnNa2(H2)10 system at 77, 200 and 300 K.
表 1 在BnNa2(n = 3—10)团簇中, Na原子的平均结合能(Eb), HOMO-LUMO能隙(Eg), Na—B平均距离(dNa—B), Na—Na平均距离(dNa—Na), Na原子的NBO电荷(QNa)
Table 1. The average binding energy (Eb) of Na atoms, HOMO-LUMO energy gap (Eg), average bond lengths of natrium-boron (dNa—B), natrium-natrium (dNa—Na) and NBO charge of Na atoms (QNa) in the BnNa2 (n = 3–10) clusters.
团簇 Eb/eV Eg/eV dNa—B/Å dNa—Na/Å QNa/e B3Na2 1.876 2.807 2.519 4.332 0.837 B4Na2 1.756 1.901 2.355 4.043 0.868 B5Na2 1.859 1.867 2.406 4.440 0.899 B6Na2 2.520 2.745 2.536 5.189 0.902 B7Na2 2.162 2.474 2.646 5.079 0.894 B8Na2 2.967 3.320 2.424 4.849 0.979 B9Na2 2.572 3.546 2.669 4.667 0.972 B10Na2 2.120 2.348 2.359 6.574 0.979  下载: 导出CSV
下载: 导出CSV
表 2 在BnNa2(H2)10 (n = 3—10)团簇中, 氢分子的平均吸附能(Eads), 氢分子的平均键长(dH—H), 氢分子与Na原子之间的平均距离(dNa—H2), Na—B平均距离(dNa—B)以及储氢密度
Table 2. The average adsorption energy (Eads), average bond lengths of hydrogen-hydrogen (dH—H), natrium-hydrogen molecule (dNa—H2), natrium-boron (dNa—B) and gravimetric density of H2 of BnNa2(H2)10 (n = 3–10) cluster.
饱和吸氢结构 Eads/eV dH—H/Å dNa—H2/Å dNa—B/Å H2/% B3Na2(H2)10 0.063 0.749 2.561 2.550 20.45 B4Na2(H2)10 0.066 0.749 2.578 2.379 18.43 B5Na2(H2)10 0.073 0.749 2.569 2.474 16.77 B6Na2(H2)10 0.085 0.749 2.552 2.672 15.39 B7Na2(H2)10 0.077 0.748 2.590 2.620 14.22 B8Na2(H2)10 0.088 0.748 2.553 2.477 13.21 B9Na2(H2)10 0.087 0.748 2.583 2.668 12.33 B10Na2(H2)10 0.095 0.749 2.535 2.433 11.57
下载: 导出CSV
表 3 BnNa2 (n = 3—10)团簇吸附氢分子前后每个原子上的平均NBO电荷
Table 3. Average NBO charges on each atom before and after H2 adsorption of BnNa2 (n = 3–10) clusters.
团簇 吸氢前 饱和吸氢后 QB/e QNa/e QB/e QNa/e QH/e B3Na2 –0.558 0.837 –0.565 0.848 0.001 B4Na2 –1.023 0.938 –0.896 0.866 0.001 B5Na2 –0.837 0.899 –0.685 0.878 0.001 B6Na2 –0.675 0.902 –0.483 0.889 0.005 B7Na2 –0.258 0.894 –0.381 0.891 0.006 B8Na2 –0.146 0.979 –0.132 0.919 0.005 B9Na2 –0.109 0.972 –0.087 0.912 0.007 B10Na2 –0.376 0.979 –0.317 0.912 0.005
下载: 导出CSV
-
[1] Shindell D T, Lee Y, Faluvegi G 2016 Nat. Clim. Change 6 503 doi: 10.1038/nclimate2935 [2] Ceran B, Mielcarek A, Hassan Q, Teneta J, Jaszczur M 2021 Appl. Energy 297 117161 doi: 10.1016/j.apenergy.2021.117161 [3] Wróbel K, Wróbel J, Tokarz W, Lach J, Podsadni K, Czerwiński A 2022 Energies 15 8937 doi: 10.3390/en15238937 [4] Okolie J A, Patra B R, Mukherjee A, Nanda S, Dalai A K, Kozinski J A 2021 Int. J. Hydrogen Energy 46 8885 doi: 10.1016/j.ijhydene.2021.01.014 [5] Ousaleh H A, Mehmood S, Baba Y F, Bürger I, Linder M, Faik A 2024 Int. J. Hydrogen Energy 52 1182 doi: 10.1016/j.ijhydene.2023.10.218 [6] https://www.energy.gov/eere/fuelcells/hydrogen-and-fuel-cell-technologies-office [2025-2-1] [7] Liu X Y, He J, Yu J X, Li Z X, Fan Z Q 2014 Chin. Phys. B 23 067303 doi: 10.1088/1674-1056/23/6/067303 [8] Mohan M, Sharma V K, Kumar E A, Gayathri V 2019 Energy Storage 1 e35 doi: 10.1002/est2.35 [9] Kumar A, Vyas N, Ojha A K 2020 Int. J. Hydrogen Energy 45 12961 doi: 10.1016/j.ijhydene.2020.03.018 [10] Tang C M, Wang Z G, Zhang X, Wen N H 2016 Chem. Phys. Lett. 661 161 doi: 10.1016/j.cplett.2016.08.064 [11] Durgun E, Ciraci S, Zhou W, Yildirim T 2006 Phys. Rev. Lett. 97 226102 doi: 10.1103/PhysRevLett.97.226102 [12] Duraisamy P D, S P M P, Gopalan P, Angamuthu A 2024 Struct. Chem. 35 681 doi: 10.1007/s11224-023-02226-9 [13] Aal S A, Alfuhaidi A K 2021 Vacuum 183 109838 doi: 10.1016/j.vacuum.2020.109838 [14] Ma L C, Wang L C, Sun Y R, Ma L, Zhang J M 2021 Physica E 128 114588 doi: 10.1016/j.physe.2020.114588 [15] Banerjee P, Pathak B, Ahuja R, Das G P 2016 Int. J. Hydrogen Energy 41 14437 doi: 10.1016/j.ijhydene.2016.02.113 [16] Satawara A M, Shaikh G A, Gupta S K, Gajjar P N 2024 Int. J. Hydrogen Energy 87 1461 doi: 10.1016/j.ijhydene.2024.09.017 [17] Muthu R N, Rajashabala S, Kannan R 2016 Renew. Energ. 85 387 doi: 10.1016/j.renene.2015.06.056 [18] Lu Q L, Huang S G, De Li Y, Wan J G, Luo Q Q 2015 Int. J. Hydrogen Energy 40 13022 doi: 10.1016/j.ijhydene.2015.08.008 [19] Tang C M, Zhang X 2016 Int. J. Hydrogen Energy 41 16992 doi: 10.1016/j.ijhydene.2016.07.118 [20] Kumar A, Ojha S K, Vyas N, Ojha A K 2022 Int. J. Hydrogen Energy 47 7861 doi: 10.1016/j.ijhydene.2021.12.091 [21] Li H R, Zhang C, Ren W B, Wang Y J, Han T 2023 Int. J. Hydrogen Energy 48 25821 doi: 10.1016/j.ijhydene.2023.03.213 [22] Olalde-López D, Rodríguez-Kessler P L, Rodríguez-Carrera S, Muñoz-Castro A 2024 Int. J. Hydrogen Energy 107 419 [23] Si L, Tang C M 2017 Int. J. Hydrogen Energy 42 16611 doi: 10.1016/j.ijhydene.2017.05.181 [24] Ruan W, Wu D L, Xie A D, Yu X G 2011 Chin. Phys. B 20 043104 doi: 10.1088/1674-1056/20/4/043104 [25] Zhang Y F, Cheng X L 2019 Physica E 107 170 doi: 10.1016/j.physe.2018.11.041 [26] Becke A D 1992 J. Chem. Phys. 96 2155 doi: 10.1063/1.462066 [27] Lee C, Yang W, Parr R G 1988 Phys. Rev. B 37 785 doi: 10.1103/PhysRevB.37.785 [28] Miehlich B, Savin A, Stoll H, Preuss H 1989 Chem. Phys. Lett. 157 200 doi: 10.1016/0009-2614(89)87234-3 [29] Ruan W, Wu D L, Luo W L, Yu X G, Xie A D 2014 Chin. Phys. B 23 023102 doi: 10.1088/1674-1056/23/2/023102 [30] Lu T, Chen F W 2012 J. Comput. Chem. 33 580 doi: 10.1002/jcc.22885 [31] Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Petersson G A, Nakatsuji H, Li X 2016 Gaussian 16 Rev. C. 01. Wallingford, CT [32] Atış M, Özdoğan C, Güvenç Z B 2007 Int. J. Quantum Chem. 107 729 doi: 10.1002/qua.21171 [33] Ye X J, Teng Z W, Yang X L, Liu C S 2018 J. Saudi Chem. Soc. 22 84 doi: 10.1016/j.jscs.2017.07.006 [34] Li Y Y, Hu Y F, Lai Q, Yuan Y Q, Huang T X, Li Q Y, Huang H B 2023 Mol. Phys. 121 e2166881 doi: 10.1080/00268976.2023.2166881 [35] Ray S S, Sahoo S R, Sahu S 2019 Int. J. Hydrogen Energy 44 6019 doi: 10.1016/j.ijhydene.2018.12.109 -
计量
- 文章访问数: 102
- HTML全文浏览数: 102
- PDF下载数: 3
- 施引文献: 0