
 首页
首页 登录
登录 注册
注册



-
2004年, 石墨烯的成功制备引发了二维材料的研究热潮[1]. 与零带隙的石墨烯相比, 有着近乎相同几何结构的锗烯, 在具有600000 cm2/(V·s)高电子迁移率[2]的同时, 自旋轨道耦合下还能打开大约24 meV的带隙[3], 但如此小的带隙同样限制了锗烯在电子与光电子器件中的应用. 理论研究表明, 表面氢化是打开锗烯带隙的有效手段. 早在2010年, Lew Yan Voon等[4–6]发现, 完全氢化的锗烯, 又称为锗烷, 是一种具有大约1.5 eV直接带隙的半导体材料, 并具有出色的物理化学性能, 在各类电子器件中可能有着广泛的应用前景. 2013年, Bianco等[7]利用CaGe2的拓扑化学脱嵌技术终于成功合成了锗烷, 其1.59 eV的直接带隙与基于HSE06(Heyd-Scuseria-Ernzerhof)的理论计算结果相符, 其18000 cm2/(V·s)的电子迁移率约为锗晶体材料的5倍. 进一步的X射线光电子能谱(X-ray photo electron spectroscopy, XPS)和傅里叶变换红外光谱(Fourier transform infrared spectroscopy, FTIR)测试表明, 锗烷还具有较好的抗氧化能力和热稳定性[8]. 基于锗烷优异的物理学化学特性, 利用锗烷制备的场效应晶体管(field effect transistor, FET)[9–11]和二极管[12]也陆续见于报道, 尤其是锗烷基的肖特基二极管相比于传统的锗二极管具有更优异的整流性能.
为进一步拓展锗烷在电子和光电子学中的应用, 研究人员通过应变[13,14]、分子吸附[15]、电场[16]等方法调控了锗烷的电子特性, 但其研究对象大多采用基于完美构型的锗烷基底, 而锗烷的实际制备过程中势必有缺陷的存在, 尤其是氢空位缺陷[17]. 因此, 研究具有氢空位缺陷的锗烷对其实际应用具有非常重要的意义. 另外, 目前较为成熟的脱氢技术, 如通过扫描隧道显微镜尖端诱导[18]的方式, 可在锗烷表面实现任意位置和任意数量的脱氢, 这也为理论与实验研究氢空位缺陷的锗烷提供了强有力的技术支撑.
作为表面氢空位缺陷的典型代表, 单氢空位缺陷极易引入锗烷表面并造成锗烷电子结构和物理化学性能的变化. 已有研究结果表明, 单氢空位缺陷的引入可使锗烷发生磁性的变化[19]. 作为调控材料特性的一种常见方法和有效手段, 双轴应变都可方便灵活地在理论与实践中得以实现, 且研究显示应变能够很好地改善材料的电子结构[20,21]. 而双轴应变下单氢空位锗烷的几何结构、电子结构与输运性质的变化, 至今为止还未开展系统深入的研究, 为此本文基于密度泛函理论的第一性原理计算方法, 研究了单氢空位锗烷的几何结构、电子结构与输运性质随双轴应变的变化情况, 发现拉伸应变至一定程度时, 可使单氢空位缺陷锗烷产生从类P型掺杂向类N型掺杂的转变, 并揭示了其内在工作机理. 进一步的输运性质计算也表明, 双轴应变可线性调控单氢空位缺陷锗烷的电子有效质量, 且拉伸应变有助于提高材料的电子迁移率与电导率.
-
本文采用基于密度泛函理论的第一性原理计算方法, 所有计算均在VASP软件包中实现[22]. 其中, 电子与离子间的相互作用可通过投影增广平面波(projected augmented wave, PAW)方法[23]描述, 电子与电子间相互作用的交换关联函数则采用广义梯度近似(generalized gradient approximation, GGA)中PBE(Perdew-Burke-Ernzerhof)[24]泛函进行处理. 模型构建中采用5 × 5 × 1的锗烷超胞(包含50个Ge原子和50个H原子), 并在Z方向上构建20 Å的真空层, 以避免层间的相互作用. 选择从锗烷中去除一个H原子来模拟单个氢空位缺陷. 计算时的平面波截断能设为500 eV, 并通过共轭梯度法对整个结构进行弛豫, 直到每个原子上的受力小于0.02 eV/Å, 而能量收敛标准设置为10–5 eV. 布里渊区采用Monkhorst-Pack[25]型网格, K点采用2 × 2 × 1的取值用于结构弛豫, 3 × 3 × 1用于电子结构计算. X和Y轴上使用的双轴应变定义为
其中,
$ a $ 和$ {a}_{0} $ 分别是有应变和无应变时锗烷的晶格常数. 输运性质的计算基于非平衡格林函数, 使用Nanodcal [26–28]软件包进行, 采用双ζ极化(double zeta polarization, DZP)原子轨道基, 截断能设为80 hartree (1 hartree = 4.356 × 10–18 J), 利用线性自旋极化PBE交换相关函数计算了不同应变下的I-V特性曲线、电子有效质量和电子迁移率. -
5 × 5 × 1锗烷超晶胞优化后的dGe—H (锗—氢键长)和dGe—Ge (锗—锗键长)分别为1.563 Å和2.467 Å, 褶皱高度Δ (即锗烷中沿Z方向上上层锗原子与下层锗原子高度的平均差值)为0.738 Å. 这些结果与前人基于密度泛函理论(density functional theroy, DFT)的研究结果高度一致[15,19,29–31]. 接着选取其中一个锗原子并去除与其相连的氢原子, 以模拟单氢空位缺陷的存在, 如图1所示(其中Ge25表示单氢空位锗烷中脱去了氢原子的锗原子, Gen表示与Ge25最近邻的锗原子, α1表示Gen—Ge25—Gen夹角, α表示非脱氢处Ge—Ge—Ge之间的平均键角). 与完美锗烷中1.563 Å的Ge—H键长相比, 单氢空位锗烷因其缺陷处的局部形变, 其Gen—H键长略微增长至1.568 Å. 其缺陷处的Ge25—Gen键长从原本的2.467 Å略微增长到2.480 Å, 相应的Ge25与最近邻Gen的键角(α1)也从111.47°减小到110.99°. 而非脱氢处部分无论是Ge—H, Ge—Ge键长还是α键角都与完美锗烷保持一致, 其褶皱高度Δ也仅从0.738 Å增至0.739 Å, 这表明单氢空位的存在对锗烷的几何结构几乎没有产生大的影响.
另外, 其自旋电荷密度图清晰地表明, 脱氢处的Ge25上存在明显的自旋电荷分布, 表现出典型的铁磁特征. 与完美锗烷相比, 这说明单氢空位锗烷已发生了磁性的转变. 除此之外, 相比于完美锗烷的电子结构(如附加材料图S1所示
(online) ), 单氢空位锗烷的自旋向上能带结构和自旋向下的能带结构明显不对称(如图2所示). 与完美锗烷0.997 eV的带隙相比, 其自旋向上能带结构的带隙为0.994 eV, 而自旋向下能带结构的带隙为1.011 eV. 更为重要的是, 在其自旋向下的能带结构中, 价带顶(valence band maximum, VBM)和导带底(conduction band minimum, CBM)之间出现了一条平直的能量色散, 通过分析投影态密度(projected density of states, PDOS)可知, 这一平直的能量色散源自于脱氢处的锗原子(Ge25), 且主要由其Pz轨道贡献. 显然, 这是因为氢原子的脱去, 导致Ge25存在孤立的不成对电子并形成悬挂键. 进一步的磁矩计算表明, 单氢空位锗烷的磁矩大小为1.000μB, 这也与上述分析及其铁磁性表现相吻合. 考虑到自旋极化作用下的能级劈裂, 缺陷态能级在自旋向上和自旋向下的能带结构中分别产生了两条具有不同能量的缺陷能级γ和γ1, 而γ1恰巧处于费米能级与其导带底之间, 从而形成了典型的受主能级, 进而产生了类P型掺杂效应. 这意味着电子从价带顶激发到非占据态的最小能量可从完美锗烷时的0.997 eV降低到现在的Ep = 0.805 eV (Ep定义为价带顶到最低非占据态分子轨道之间的能量差). 这种由单个氢缺陷引起的类P型的掺杂效应可能也会像传统半导体掺杂剂引起的掺杂效应一样, 影响锗烷的电子输运特性, 而如果能有效地调控这种掺杂效应的同时并改善材料的电子结构, 或许能使其更好地应用于相关领域. 已有研究成果表明, 双轴应变是一种灵活调控材料电子特性的有效方法, 如Li和Chen[13]发现双轴应变能够很好地调控锗烷的带隙, 而Yan等[14]也使用双轴应变的方法改变了锑烯/锗烷异质结的能带结构, 提高了材料的电子迁移率. 因此, 双轴应变成为调控时优先考虑的方法, 综合实验中所能产生的实际应变大小, –3%—3%的双轴应变被用来进一步研究其对单氢空位锗烷几何结构、电子结构和输运性质的调控. -
表1所示为锗烷脱氢处和非脱氢处在–3%—3%双轴应变下键长、键角及褶皱高度的变化. 当应变从–3%—3%变化时, 其Gen—H和非脱氢处Ge—H键长分别从1.566 Å 和1.559 Å增至1.571 Å 和1.566 Å; 与无应变时的1.568 Å和1.563 Å相比, 双轴应变下的Ge—H键长变化很小. 而Ge25—Gen键长和Ge—Ge键长在此应变范围内, 可从–3%时的2.432 Å和2.417 Å分别拉伸至3%时的2.545 Å和2.522 Å. 对应键角α, α1的变化范围则分别为109.85°—112.83°和109.36°—112.20°. 值得注意的是, 锗烷的褶皱高度对应变同样敏感, 在–3%—3%双轴应变下, 其褶皱高度可从0.791 Å降至0.695 Å. 为更清晰地描述单氢空位锗烷几何结构随应变的变化规律, 图3所示为单氢空位锗烷键长、键角及褶皱高度随应变的变化趋势. 很明显, 除了Gen—H和Ge—H键长几乎不受应变影响外, Ge25—Gen键长、Ge—Ge键长、键角α, α1和褶皱高度Δ几乎均随应变呈线性变化, 这表明双轴应变可线性调控单氢空位锗烷的几何结构.
-
研究发现, 锗烷的带隙极易受双轴应变线性的调控[13], 而单氢空位的缺陷态能量也有可能因为双轴应变下体系褶皱高度的变化, 导致体系费米能级和杂化轨道的能量变化, 使得缺陷态掺杂效应的强弱和类型发生变化. 为探索不同应变下掺杂效应的变化, 图4和图5分别描述了单氢空位锗烷在0%—3%的双轴压缩应变和双轴拉伸应变下的能带结构. 由图4(a)—(d)可知, 在0%—3%的压缩应变范围内, 体系自旋向下能带结构中的缺陷带随着压缩应变强度增大逐渐靠近费米能级, 相应的Ep从0.805 eV逐渐减小到0.440 eV. 而随着压缩应变强度的增大, 导带底逐渐远离费米能级, 因价带顶并未发生明显变化, 导致其带隙Eg也从1.010 eV增大到1.465 eV, 这表明通过双轴压缩应变可以增大Eg并减小Ep. 而Ep的减小代表着类P型掺杂效应的增强, 即降低了电子从价带顶激发到非占据态所需的能量.
从Ep与CBM随压缩应变的变化趋势来看, 随着压缩应变的减小, 自旋向下能带结构中的缺陷带能级逐渐远离费米能级, 而CBM却逐渐靠近费米能级, 这意味着有可能在某一个临界点处, 缺陷带能级恰好与导带底能量相同, 即类P型掺杂效应可能消失. 为验证这一猜想, 在0%—3%的拉伸应变中采用二分的步进方法进行搜索, 确实在拉伸应变为0.75%处发现了这一临界点, 如图5(b)所示. 即当拉伸应变为0.75%时, Ep达到最大值0.826 eV, 图S2
(online) 的能带结构图和投影态密度(PDOS) 图也清晰地表明, 当拉伸应变为0.75%时, 缺陷态的峰与导带底的峰的确处于同一能级, 进一步证明了当拉伸应变为0.75%时, 类P型掺杂效应即将消失. 而随着拉伸应变的增大, 自旋向下的能带结构中缺陷态能级进一步远离费米能级而导带底进一步靠近费米能级, 如图5(c), (d)所示. 此时, 自旋向上的能带结构中, 缺陷态能级却在逐渐靠近价带顶与费米能级. 可以判断, 随着拉伸应变的进一步增大, 自旋向上能带结构中缺陷态能级有可能会超过价带顶, 从而在自旋向上的能带结构中形成施主能级, 从而形成类N型的掺杂效应. 同样的二分步进方法在拉伸应变达到2.5%时发现, 自旋向上能带结构中的缺陷能带恰好与价带顶相切, 且由缺陷带提供的峰与价带顶提供的峰在同一能级(如图5(e)和图S3 (online )所示), 这表明其电子结构中的确出现了类N型掺杂效应. 此时缺陷态能级作为施主能级, 产生的类N型掺杂效应的En = 0.623 eV (定义最高占据态分子轨道到导带底之间的能量差为En). 当拉伸应变进一步增至3%时, En和Eg的值都相应地减小到0.519 eV和0.568 eV, 这表明拉伸应变可有效减小En和Eg.为探索应变对单氢空位锗烷电子结构和掺杂效应的调控机理, 我们计算了–3%—3%双轴应变下单氢空位锗烷的分轨道投影态密度, 如图6(a)所示. 图6(a)中无应变时自旋向上的Pz轨道能级约为0.4 eV, 自旋向下的Pz轨道能级约为0.6 eV, 这与图4(a)中无应变下的两个缺陷能级大小保持一致. 而主要由S轨道贡献的导带底能级大小也与PDOS中S轨道能级大小一致. –3%—3%应变下Pz轨道和S轨道变化趋势可与图4和图5中缺陷能级和导带底能级变化趋势一一对应, 所以从相对费米能级的角度上来说, 主要是由于Pz轨道提供的缺陷能级和S轨道提供的导带底能级随着应变发生相应的变化, 导致不同的掺杂效应类型、程度以及带隙的变化.
为分析其电子结构变化的内在机理, 本文进一步计算了费米能级(E-fermi)、导带底(CBM)、价带顶(VBM)、最高占据态分子轨道(highest occupied molecular orbital, HOMO)和最低非占据态分子轨道(lowest unoccupied molecular orbital, LUMO)相对于真空能级的绝对能量变化, 结果如图6(b)所示. 发现双轴应变下, 自旋向上和自旋向下的HOMO和LUMO的绝对能量变化并不太明显, 这是因为脱氢处Ge原子上的局域电荷即使在双轴应变下仍可保持相对稳定, 这也与前人文献中的结果相符合[32]. 而在0%—3%双轴压缩应变下, E-Fermi, CBM和VBM的绝对能量均为线性上升的, 且VBM与费米能级的上升速率几乎保持一致, 因此价带顶相对于费米能级的能量变化很小, 故图4和图5中价带顶相对于费米能级几乎维持不变. 对CBM而言, 其随应变线性上升的速率超过了费米能级和VBM的线性上升速率. 这一现象可以通过轨道杂化理论作出合理的解释与分析. 如图7(a)—(c)所示, 随着拉伸应变的增大, 单氢空位锗烷褶皱高度Δ随之下降, 而Ge—Ge键长随之变长, 体系结构逐渐趋向sp2转变, 促使其spz杂化轨道上的反键态能量降低, 电荷更多地局域在Ge原子顶部或底部, 导致主要由spz杂化轨道贡献的CBM绝对能量显著降低. 与之相对应的主要由px, py轨道贡献的VBM能量, 则随着拉伸应变导致的面内轨道杂化作用的减弱, 也逐渐降低, 但局域在Ge—Ge键上的电荷变化相对于CBM的显著变化而言较为平缓, 如图8(a)—(c)所示. 因此, VBM绝对能量的变化速率小于CBM的变化速率, 故整体而言体系的带隙随着拉伸应变的增大在线性降低, 如图6(c)所示. 尤其当应变为0.75%时, CBM的能量与LUMO的能量相同, 造成了类P型掺杂效应在此处的临界点; 而随着VBM的缓慢下降, 当应变为2.50%时, VBM的能量与HOMO的能量相同, 造成了此处类N型掺杂效应临界点的出现.
值得注意的是, 由此造成的Ep在–3%—0.75%双轴应变下, 呈现出随应变线性变化的规律, 如图6(c)所示. 故–3%—3%的双轴应变不仅能线性地调控体系带隙, 且可在不同的应变下出现不同的体系掺杂效应. 其内在工作机理正是因为应变改变了单氢空位锗烷的褶皱高度和Ge—Ge键长变化, 从而改变了E-Fermi, CBM, VBM的能量并引起缺陷态能级的相对移动, 从而造成可调控的电子结构与掺杂效应.
-
为探索–3%—3%双轴应变下单氢空位锗烷的电子输运特性, 并考虑锗烷沿Armchair和Zigzag两种不同的输运方向, 在Device Studio软件平台上构建了由左右电极和中心区域组成的输运模型, 如附加材料图S4
(online) 所示. 通过对左右两侧电极施加0—2.6 V的偏置电压, 计算了单氢空位锗烷在–3%—3%双轴应变下体系的电流-电压特性曲线, 如图9所示. 可以看出, 体系沿Armchair和Zigzag两种不同的输运方向上的响应几乎是重叠的, 这说明单氢空位锗烷与完美锗烷一样, 具有良好的各向同性. 当体系无应变作用时, 0—1.0 V偏压下的电流几乎为零; 而当电压>1.0 V时, 电流开始出现明显的增长. 随着电压的增大, 响应电流表现出良好的单调增长关系, 最终可在2.6 V电压下得到将近100 μA的电流. 在–1%的应变下, 其开关电压增至1.2 V左右, 而2.6 V偏压下的电流降为70 μA左右; 进一步增大压缩应变至2%和3%时, 其开关电压分别增大至1.4 V和1.6 V左右, 相应2.6 V偏压下的电流则分别降至50 μA和30 μA左右. 这表明压缩应变的增大可提高其开关电压并降低其在相同偏压下的电流. 当双轴应变为1%, 2%和3%时, 其开关电压分别降至0.9 V, 0.7 V, 0.6 V左右, 而2.6 V偏压下的电流则分别增至140, 180, 220 μA左右. 其开关电压与应变的对应关系与带隙随应变的变化规律相同, 说明带隙的变化反映了开关电压的变化.很明显, –3%—3%双轴应变下的I-V曲线具有不同的斜率, 这意味着双轴应变可改变体系的电子电导. 通过公式
$ G=\dfrac{\Delta I}{\Delta V} $ 和$ \sigma =\dfrac{GL}{S} $ (其中$ G $ 为电导,$ \sigma $ 为电导率,$ L $ 表示器件的长度,$ S $ 表示截面积)可计算得到单氢空位锗烷在–3%—3%双轴应变下的电导和电导率. 当ε = 0%时, 电导和电导率分别为69 μS 和2165 S/cm; 当ε = 3%时, 体系的电导和电导率可分别增至116 μS和3660 S/cm; 当ε = –3%时, 体系的电导和电导率可分别降至35 μS和1069 S/cm, 且随应变几乎是呈线性相关的, 如图9(b)所示. 一般来说, 半导体中的电子迁移率正比于电导率而反比于电子有效质量. 通过计算电子迁移率和电子有效质量发现, 体系的电子迁移率在–3%—3%的双轴应变下具有与电导率几乎相同的变化规律, 而电子有效质量呈现出相反的变化规律, 如图9(c)所示. 这意味着压缩应变的增大可线性增大电子有效质量而降低电子迁移率; 相反, 拉伸应变的增大可线性降低电子有效质量而提高电子迁移率. 当ε = –3%时, 电子有效质量可增至0.115m0, 相应的电子迁移率可降至14148 cm2/(V·s); 当ε = 3%时, 电子有效质量可降至0.067m0, 相应的电子迁移率可增至24252 cm2/(V·s). 同样的调控方法和变化规律也在单层过渡金属硫化物中进行过报道[33–36]. -
综上所述, 本文通过第一性原理计算方法, 研究了单氢空位对锗烷电子结构的影响, 发现单氢空位缺陷的引入可在锗烷中产生类P型掺杂效应, 而双轴应变可有效调控其掺杂效应的强度甚至发生掺杂类型的转变. 其调控机理表明, 体系VBM受应变影响较小, 但应变明显改变了缺陷态和CBM对费米能级的相对能量, 使得缺陷态在不同应变作用时表现为受主能级或施主能级, 从而形成受控于双轴应变的掺杂强度甚至是掺杂类型. 而双轴应变下的量子输运计算结果表明, 单氢空位锗烷的电子输运具有各向同性, 其门限电压受控于体系带隙且应变可线性地调控电子有效质量并改善体系的电子迁移率, 这为后续实验利用单氢空位缺陷和双轴应变调控锗烷的电子结构与输运特性提供了理论基础, 有望在未来以锗烷基的电子与光电子器件中得以应用.
应变对单氢空位锗烷电子结构和输运性质的调控
Regulation of structural, electronic, and transport properties of single hydrogen vacancy germanane by strain
-
摘要: 本文利用基于密度泛函理论的第一性原理计算方法, 研究了双轴应变对单氢空位锗烷电子结构及其输运特性的调控. 研究结果发现, 单氢空位缺陷态的引入不仅可在锗烷中产生类P型掺杂效应, 还可使锗烷发生无磁性到铁磁性的转变. –3%—3%双轴应变作用下, 单氢空位锗烷的键长、键角和褶皱高度与带隙均随应变呈线性变化; 当ε = 0.75%时, 类P型掺杂效应消失, 而进一步增大应变至ε = 2.5%时, 产生了类N型掺杂效应. 其机理分析表明, 双轴应变主要改变了费米能级、价带顶和导带底的能量, 使缺陷态能级发生了相对位置的移动, 使之成为受主能级或施主能级, 并产生受控于双轴应变的掺杂效应变化. 进一步的输运特性计算表明, 具有各向同性的单氢空位锗烷的I-V特性与电子有效质量也可线性的受控于双轴应变, 并导致其电子迁移率随之变化. 当ε = 3%时, 单氢空位锗烷的电导率与电子迁移率可分别增至3660 S/cm和24252 cm2/(V·s).Abstract: The regulation of the electronic structure and transport properties of single-hydrogen-vacancy germanane by biaxial strain is investigated using first-principles calculations based on density functional theory in this work. The results reveal that the introduction of single-hydrogen-vacancy defect states not only induces P-type doping-like effects in germanane but also triggers off a transition from non-magnetic to ferromagnetic states. Under –3% to 3% biaxial strain, both the structural parameters (bond length, bond angle, and corrugation height) and the bandgap of single-hydrogen-vacancy germanane linearly vary with strain. The P-type doping-like effect disappears at ε = 0.75%, while an N-type doping-like effect appears when strain increases to ε = 2.5%. Mechanism analysis reveals that biaxial strain primarily modulates the energies of the Fermi level, valence band maximum, and conduction band minimum, causing the relative position of defect state energy levels to shift, making them become acceptor or donor energy levels, and producing doping effect changes regulated by biaxial strain. Transport property calculations further demonstrate that the isotropic I-V characteristics and electron effective mass of single-hydrogen-vacancy germanane can be linearly controlled by biaxial strain, leading to corresponding changes in electron mobility. At ε = 3%, the electrical conductivity and electron mobility of single-hydrogen-vacancy germanane increase significantly to 3660 S/cm and 24252 cm2/(V·s), respectively.
-
Key words:
- strain /
- single hydrogen vacancy /
- electronic structure /
- transport properties .
-

-
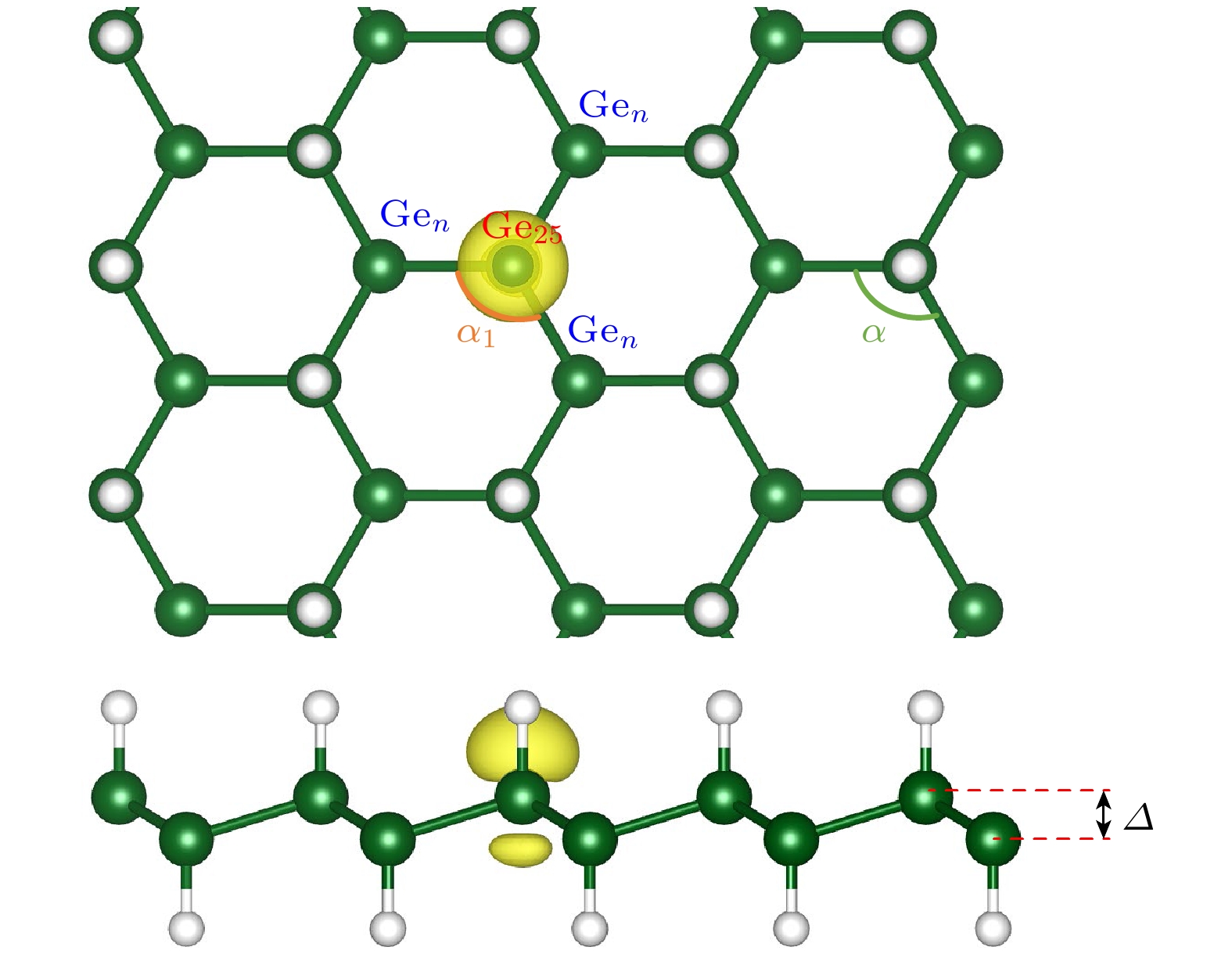
图 1 单氢空位锗烷自旋电荷密度的俯视图和侧视图(绿色和白色球体分别表示锗和氢原子, 等值面设置为0.005 Å–3)
Figure 1. Top and side views of spin charge density of single hydrogen vacancy germanane (green and white spheres represent Ge and H atoms, respectively, the isosurface is set to be 0.005 Å–3).
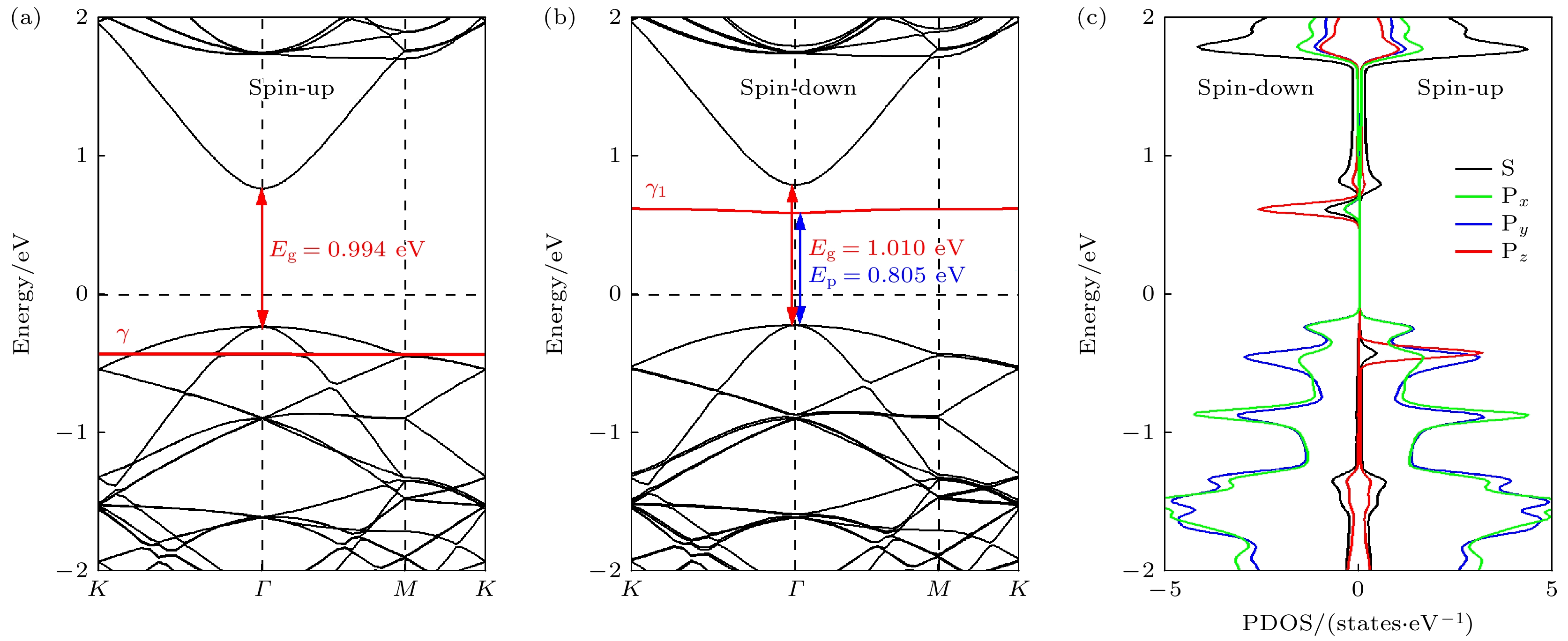
图 2 单氢空位锗烷的能带结构和投影态密度
Figure 2. Band structure and projected density of states of single hydrogen vacancy germanane.
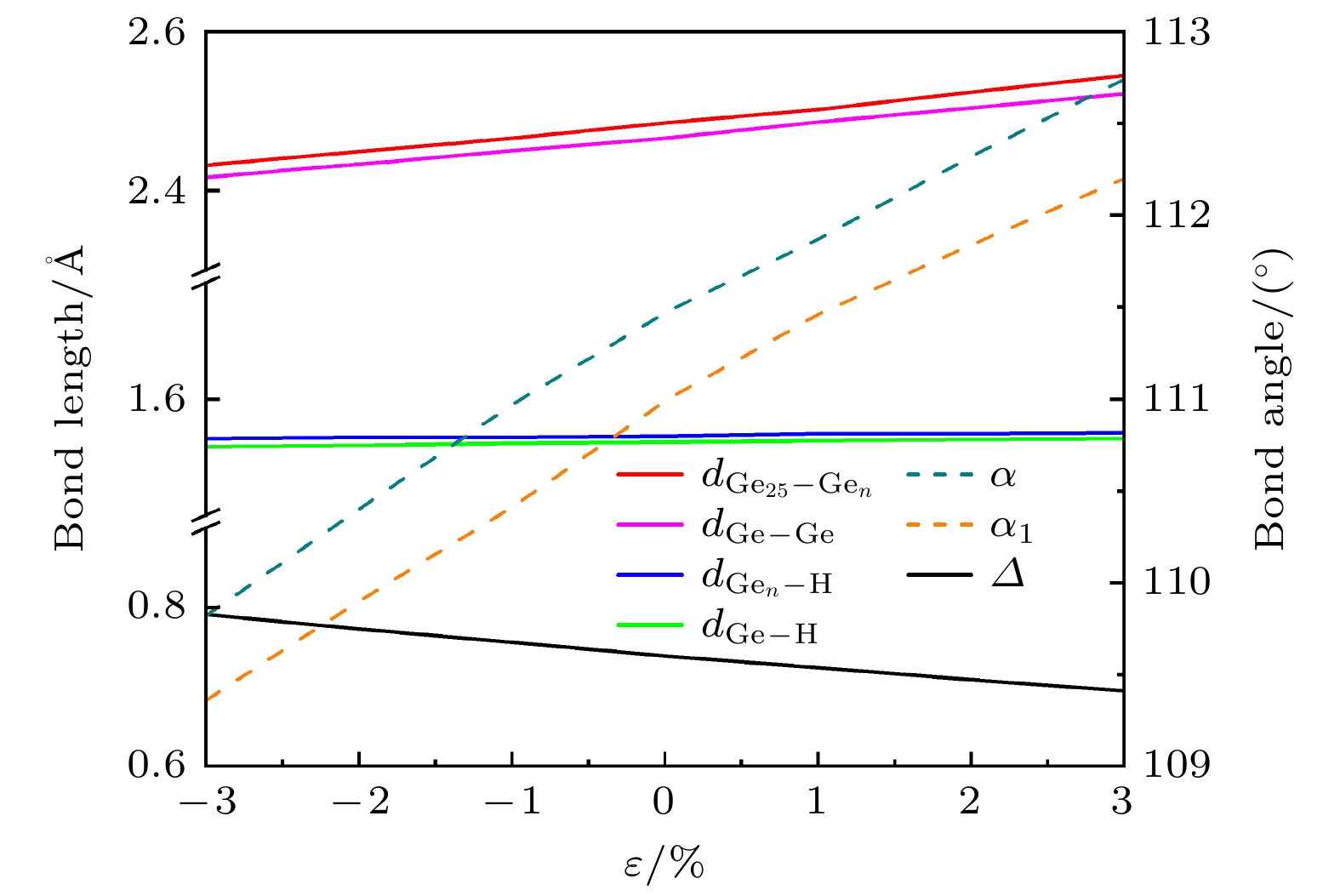
图 3 在–3%—3%双轴应变下单氢空位锗烷键长、键角和平均褶皱高度Δ的变化趋势
Figure 3. The variation trend of bond length, bond angle, and average bending height Δ of single hydrogen vacancy germanane under biaxial strain of –3% to 3%.
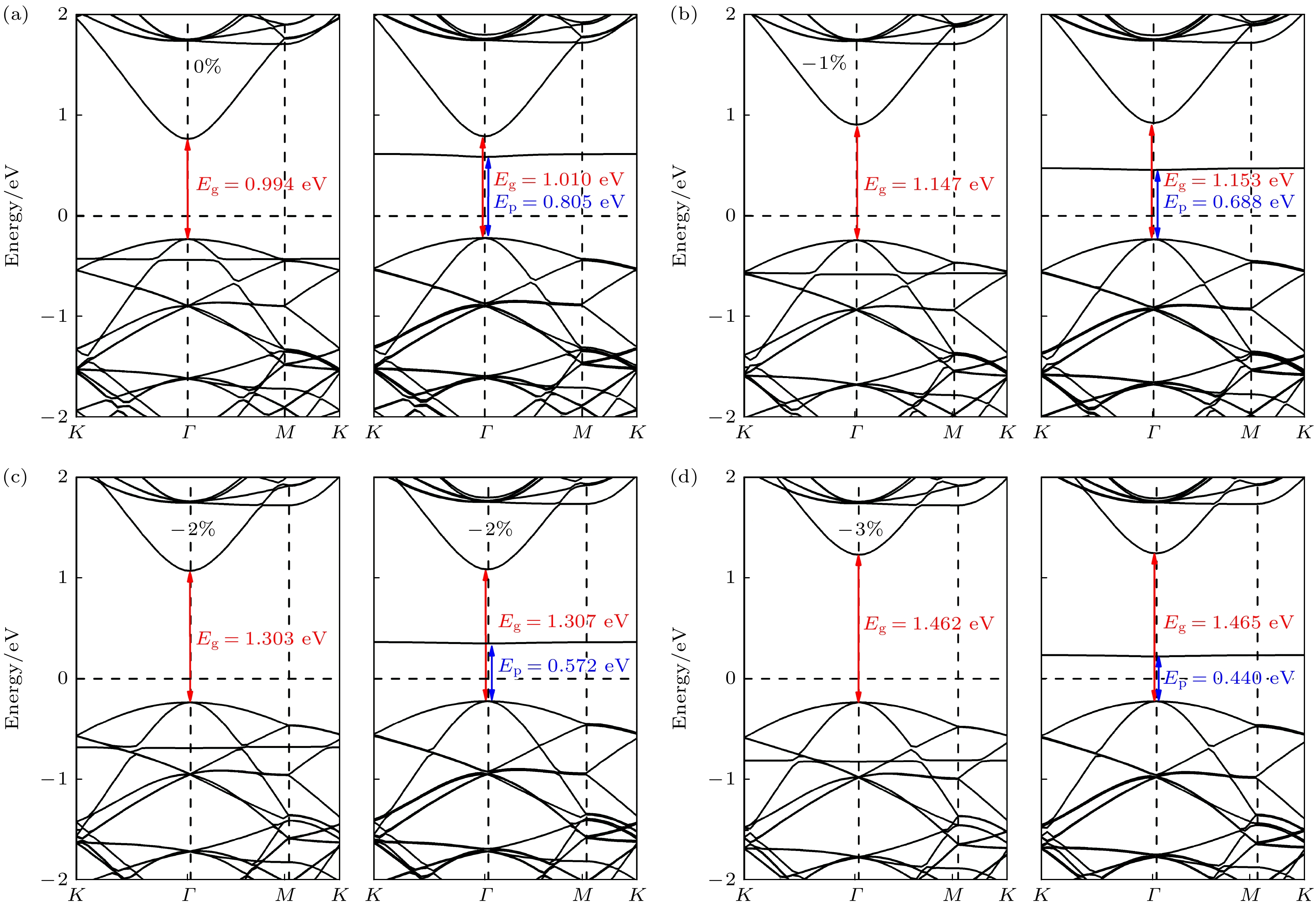
图 4 (a)—(d) 在0%—3%的压缩应变下, 单氢空位锗烷的能带结构
Figure 4. (a)–(d) The band structures of single hydrogen vacancy germanane under compression strain of 0% to 3%.
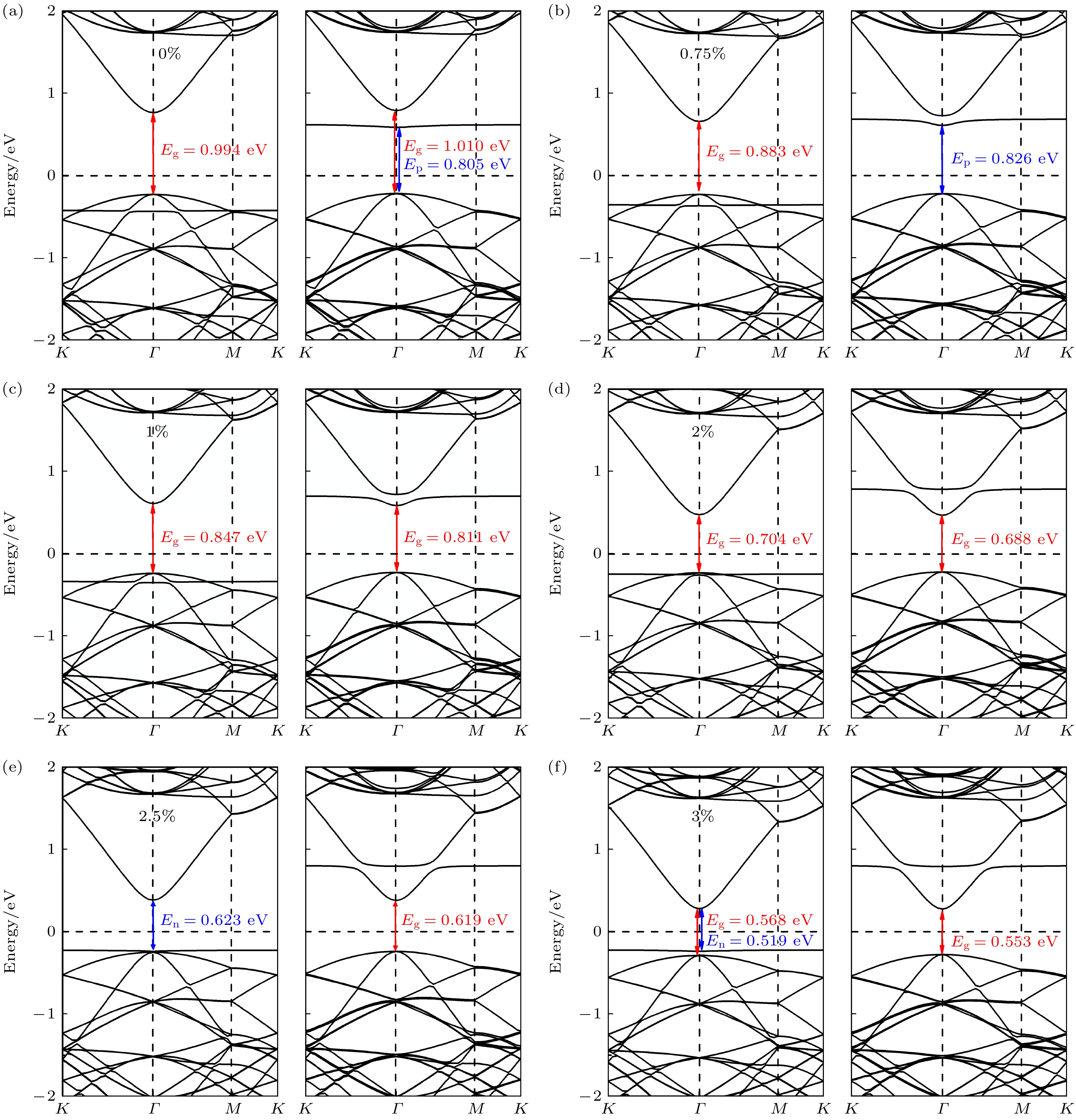
图 5 (a)—(f) 在0%—3%的拉伸应变下, 单氢空位锗烷的能带结构
Figure 5. (a)–(f) The band structures of single hydrogen vacancy germanane under tensile strain of 0% to 3%.

图 6 在–3%—3%的双轴应变下, 单氢空位锗烷 (a) 投影态密度; (b) E-fermi, CBM, VBM, HOMO和LUMO的绝对能量相对于真空能级的变化; (c) Eg, En和Ep的变化
Figure 6. Single hydrogen vacancy germanane under biaxial strain of –3% to 3%: (a) The projected density of states; (b) the evolution of absolute energy of E-fermi, CBM, VBM, HOMO and LUMO with respect to the vacuum level; (c) the evolution of the Eg, En and Ep.

图 7 在Γ点处CBM的能带分解电荷密度图(俯视图和侧视图) (a) ε = –3%; (b) ε = 0%; (c) ε = 3%
Figure 7. The band decomposed charge density of the CBM at the Γ-point (top and side views): (a) ε = –3%; (b) ε = 0%; (c) ε = 3%.
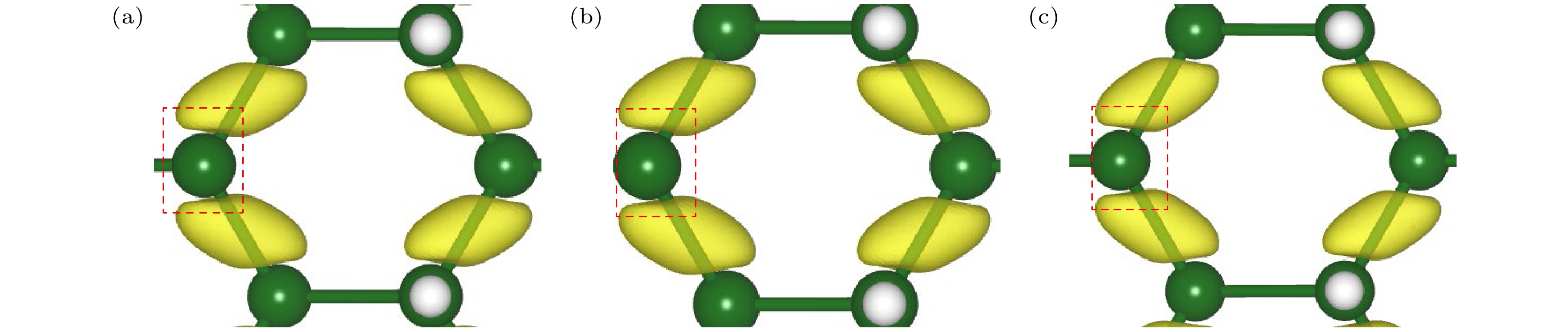
图 8 在Γ点处VBM的能带分解电荷密度图(俯视图) (a) ε = –3%; (b) ε = 0%; (c) ε = 3%
Figure 8. The band decomposed charge density of the VBM at the Γ-point (top views): (a) ε = –3%; (b) ε = 0%; (c) ε = 3%.
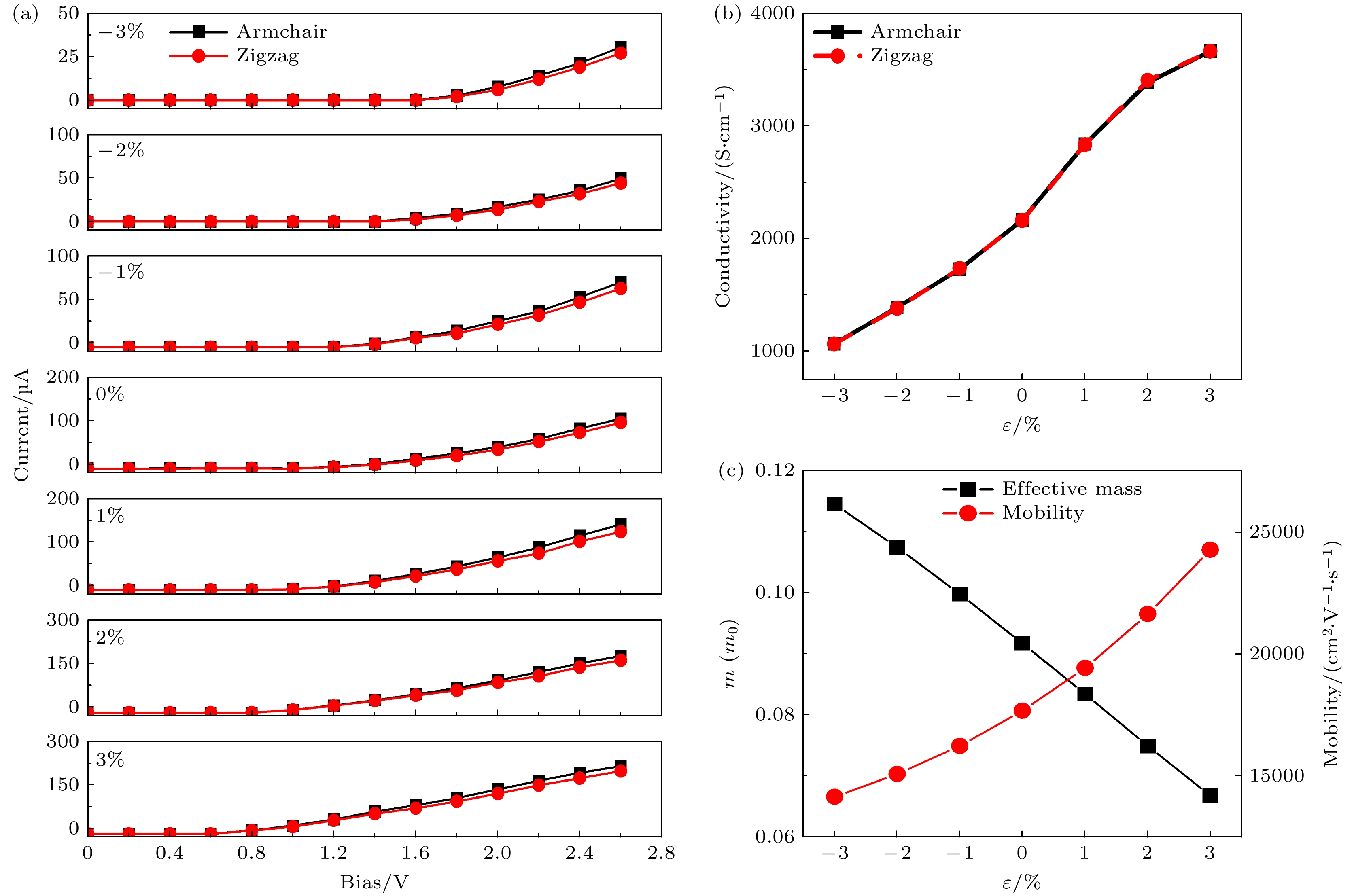
图 9 单氢空位锗烷器件在–3%—3%的双轴应变下 (a) I-V特性曲线; (b) 电导率; (c) 电子有效质量和电子迁移率
Figure 9. Single hydrogen vacancy germanane devices under biaxial strain of –3% to 3%: (a) I-V characteristic curve; (b) conductivity; (c) electronic effective mass and electronic mobility.
表 1 单氢空位锗烷的键长、键角和平均褶皱高度Δ随–3%—3%双轴应变的变化
Table 1. The variation of bond length, bond angle, and average fold height Δ of single hydrogen vacancy germanane with –3%–3% biaxial strain.
Strain dGe—H/Å dGe—Ge/Å α/(°) $d_{{\rm Ge}_n—{\rm H}}$ /Å$d_{{\mathrm{Ge}}_{25}—{\mathrm{Ge}}_{n}} $ /Åα1/(°) Δ/Å –3% 1.559 2.417 109.85 1.566 2.432 109.36 0.791 –2% 1.560 2.433 110.42 1.567 2.449 109.90 0.773 –1% 1.562 2.450 111.00 1.567 2.466 110.41 0.756 0% 1.563 2.466 111.47 1.568 2.485 110.99 0.739 1% 1.564 2.486 111.90 1.570 2.502 111.46 0.724 2% 1.565 2.504 112.39 1.570 2.524 111.84 0.709 3% 1.566 2.522 112.83 1.571 2.545 112.20 0.695  下载: 导出CSV
下载: 导出CSV
-
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I V, Firsov A A 2004 Science 306 666 doi: 10.1126/science.1102896 [2] Ye X S, Shao Z G, Zhao H B, Yang L, Wang C L 2014 RSC Adv. 4 21216 doi: 10.1039/C4RA01802H [3] Liu C C, Jiang H, Yao Y 2011 Phys. Rev. B 84 195430 doi: 10.1103/PhysRevB.84.195430 [4] Lew Yan Voon L C, Sandberg E, Aga R S, Farajian A A 2010 Appl. Phys. Lett. 97 163114 doi: 10.1063/1.3495786 [5] Houssa M, Pourtois G, Afanas’ev V V, Stesmans A 2010 Appl. Phys. Lett. 96 082111 doi: 10.1063/1.3332588 [6] Houssa M, Scalise E, Sankaran K, Pourtois G, Afanas’ev V V, Stesmans A 2011 Appl. Phys. Lett. 98 223107 doi: 10.1063/1.3595682 [7] Bianco E, Butler S, Jiang S, Restrepo O D, Windl W, Goldberger J E 2013 ACS Nano 7 4414 doi: 10.1021/nn4009406 [8] Jiang S, Butler S, Bianco E, Restrepo O D, Windl W, Goldberger J E 2014 Nat. Commun. 5 3389 doi: 10.1038/ncomms4389 [9] Xu L Y, Liu J C, Shao C, Li H, Ma W Q, Yan J F, Zhang Y Y, Dai Y, Lei X Y, Liao C G, Zhang Z Y, Zhao W, Lu J, Zhang H 2024 J. Appl. Phys. 135 134303 doi: 10.1063/5.0192389 [10] AlMutairi A, Zhao Y, Yin D, Yoon Y 2017 IEEE Electron Device Lett. 38 673 doi: 10.1109/LED.2017.2681579 [11] Zhao Y, AlMutairi A, Yoon Y 2017 IEEE Electron Device Lett. 38 1743 doi: 10.1109/LED.2017.2763120 [12] Sahoo N G, Esteves R J, Punetha V D, Pestov D, Arachchige I U, McLeskey J T 2016 Appl. Phys. Lett. 109 023507 doi: 10.1063/1.4955463 [13] Li Y F, Chen Z F 2014 J. Phys. Chem. C 118 1148 doi: 10.1021/jp411783q [14] Yan J, Cao D, Yang X, Wang J F, Jiang Z T, Jiao Z W, Shu H B 2022 Appl. Phys. A 128 958 doi: 10.1007/s00339-022-06093-9 [15] Wang X, Liu G, Liu R F, Luo W W, Wu M S, Sun B Z, Lei X L, Ouyang C Y, Xu B 2018 Nanotechnology 29 465202 doi: 10.1088/1361-6528/aae046 [16] Ye J P, Liu G, Han Y, Luo W W, Sun B Z, Lei X L, Xu B, Ouyang C Y, Zhang H L 2019 Phys. Chem. Chem. Phys. 21 20287 doi: 10.1039/C9CP04122B [17] Chen Q, Liang L, Potsi G, Wan P, Lu J, Giousis T, Thomou E, Gournis D, Rudolf P, Ye J 2019 Nano Lett. 19 1520 doi: 10.1021/acs.nanolett.8b04207 [18] Qiu J, Wang H, Wang J, Yao X, Meng S, Liu Y 2022 Phys. Rev. B 106 184102 doi: 10.1103/PhysRevB.106.184102 [19] Zhao J, Zeng H 2016 RSC Adv. 6 28298 doi: 10.1039/C5RA23323B [20] Wang X, Liu G, Liu R F, Luo W W, Sun B Z, Lei X L, Ouyang C Y, Xu B 2019 J. Appl. Phys. 125 082504 doi: 10.1063/1.5050943 [21] Zeng J C, Liu G, Han Y, Luo W W, Wu M S, Xu B, Ouyang C Y 2021 ACS Omega 6 14639 doi: 10.1021/acsomega.1c01747 [22] Kresse G, Hafner J 1993 Phys. Rev. B: Condens. Matter 47 558 doi: 10.1103/PhysRevB.47.558 [23] Blochl P E 1994 Phys. Rev. B: Condens. Matter 50 17953 doi: 10.1103/PhysRevB.50.17953 [24] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 doi: 10.1103/PhysRevLett.77.3865 [25] Monkhorst H J, Pack J D 1976 Phys. Rev. B 13 5188 doi: 10.1103/PhysRevB.13.5188 [26] Taylor J, Guo H, Wang J 2001 Phys. Rev. B 63 245407 doi: 10.1103/PhysRevB.63.245407 [27] Brandbyge M, Mozos J L, Ordejón P, Taylor J, Stokbro K 2002 Phys. Rev. B 65 165401 doi: 10.1103/PhysRevB.65.165401 [28] Yang W, Cao Y, Han J C, Lin X H, Wang X H, Wei G D, Lv C, Bournel A, Zhao W S 2021 Nanoscale 13 862 doi: 10.1039/D0NR07290G [29] Hu L, Zhao J, Yang J L 2014 J. Phys. Condens. Matter 26 335302 doi: 10.1088/0953-8984/26/33/335302 [30] Liu L, Ji Y J, Liu L Q 2019 Bull. Mater. Sci. 42 157 doi: 10.1007/s12034-019-1843-z [31] 杨子豪, 刘刚, 吴木生, 石晶, 欧阳楚英, 杨慎博, 徐波 2023 物理学报 72 127101 doi: 10.7498/aps.72.20230170 Yang Z H, Liu G, Wu M S, Shi J, Ouyang C Y, Yang S B, Xu B 2023 Acta Phys. Sin. 72 127101 doi: 10.7498/aps.72.20230170 [32] Zhou Y G, Liu K Z, Xiao H Y, Xiang X, Nie J L, Li S A, Huang H, Zu X T 2015 J. Mater. Chem. A 3 3128 [33] Tong X Y, Fang L, Liu R L 2019 AIP Adv. 9 055324 doi: 10.1063/1.5091705 [34] Chung Y F, Chang S T 2024 Nanomaterials 14 1420 doi: 10.3390/nano14171420 [35] Yu D C, Zhang Y, Liu F 2008 Phys. Rev. B 78 245204 doi: 10.1103/PhysRevB.78.245204 [36] Hosseini M, Elahi M, Pourfath M, Esseni D 2015 J. Phys. D: Appl. Phys. 48 375104 doi: 10.1088/0022-3727/48/37/375104 -
计量
- 文章访问数: 32
- HTML全文浏览数: 32
- PDF下载数: 1
- 施引文献: 0