
 首页
首页 登录
登录 注册
注册



-
锂离子电池在新能源存储方面具有十分重要的地位, 然而锂离子电池在价格、安全与性能方面存在的不足, 使非锂离子电池的研究与开发成为近年来的热门课题. 但非锂离子电池的探索也存在一些难题, 例如: Na+和K+因其尺寸过大而导致扩散动力学减弱[1,2], 而镁离子电池则存在可逆性较差的问题[3,4]. 为了解决目前所存在的问题, 研究开发新型阳极材料势在必行. 二维材料因其独特的结构特点以及优异的电化学性能在金属离子电池的应用领域获得了广泛的关注. 由此, 开发具有高性能的二维阳极材料对金属离子电池的发展至关重要[5].
优异的金属离子电池阳极材料需要具备较高的理论容量、较低的扩散势垒以及较小的体积变化率. 在近年来的研究中, 含有Be, B元素的二维材料在金属离子电池阳极的应用方面拥有出色的表现, 因其较小的摩尔质量, 在作为阳极时表现出较高的理论容量. Mortazavi等[6]研究证明了硼烯作为镁离子电池阳极时拥有较高的理论容量(2480 mA·h·g–1)且对Mg离子表现出较强的吸附性. Yeoh等[7]研究发现单层Be2C作为锂离子电池阳极材料时所表现出的高性能, 即较高的理论容量(1785 mA·h·g–1)和较低的低扩散势垒(0.11 eV). Saif等[8]研究了BeC7作为锂离子电池阳极时其理论容量可达2303.295 mA·h·g–1. Ye等[9]对Be2B进行研究, 发现这种材料具有稳定的对镁离子吸附的能量(–0.7 eV)、超高的理论容量(7436 mA·h·g–1)以及较低的扩散势垒(0.1—0.37 eV). Wan等[10]系统地评估了BeB2的性能, 并认为其具有较高理论容量(1785 mA·h·g–1)、较低的平均开路电压(0.33 V)和扩散势垒(0.11 eV), 是作为锂离子电池阳极的优秀材料. Wu和Hou[11]对Be2B2进行了深入的研究, 发现这种材料作为锂离子电池电极时具有较高的理论容量(1352 mA·h·g–1)和较低的扩散势垒(0.36 eV)的特性, 并预测该种材料将在未来具有很广阔的应用前景[11]. 根据以上研究, 可以得知Be基和B基作为电池电极时通常会表现出较高的理论容量和低扩散势垒, 由此我们有理由认为此类材料具有较好的发展前景. 本文采用基于密度泛函理论的第一性原理计算来展开对二维BeB2作为镁离子电池阳极的深入研究. 根据计算, 我们认为BeB2是一种稳定材料, 在吸附镁离子后其金属性保持不变, 而镁离子也表现了各向异性的扩散特征. 此外, 二维BeB2展示了超高的理论容量和较低的开路电压, 在镁离子电池阳极的应用方面表现出色.
-
本文所有计算均基于密度泛函理论的第一性原理计算展开, 利用Material Studio中的Cambridge Sequential Total Energy Package (CASTEP)模块来完成相关计算[12]. 电子交换关联泛函选用广义梯度近似(GGA)近似下的Perdew-Burke-Ernzerhof (PBE)泛函[13]. 在结构优化计算中, 原子弛豫的能量阈值为10–5 eV·atom–1, 力的收敛标准为0.01 eV·Å–1. 平面波的计算采用超软赝势的方法, 截止能量设为520 eV. 采用TS(Tkatchenko and Scheffler)提出的方案描述范德瓦耳斯(vdW)相互作用[14,15]. 同时, c 方向上的真空层厚度设置为 20 Å, 以减轻相邻单层结构之间的相互作用. 布里渊区K点的采样密度设置为14×14×1. 在Mg的吸附计算过程中, 采用3×3×1的BeB2超级原胞作为基底. 根据LST/QST方法计算 Mg2+在 BeB2表面的扩散势垒及过渡态数据[16], 同时运用NEB方案确认过渡态[17].
-
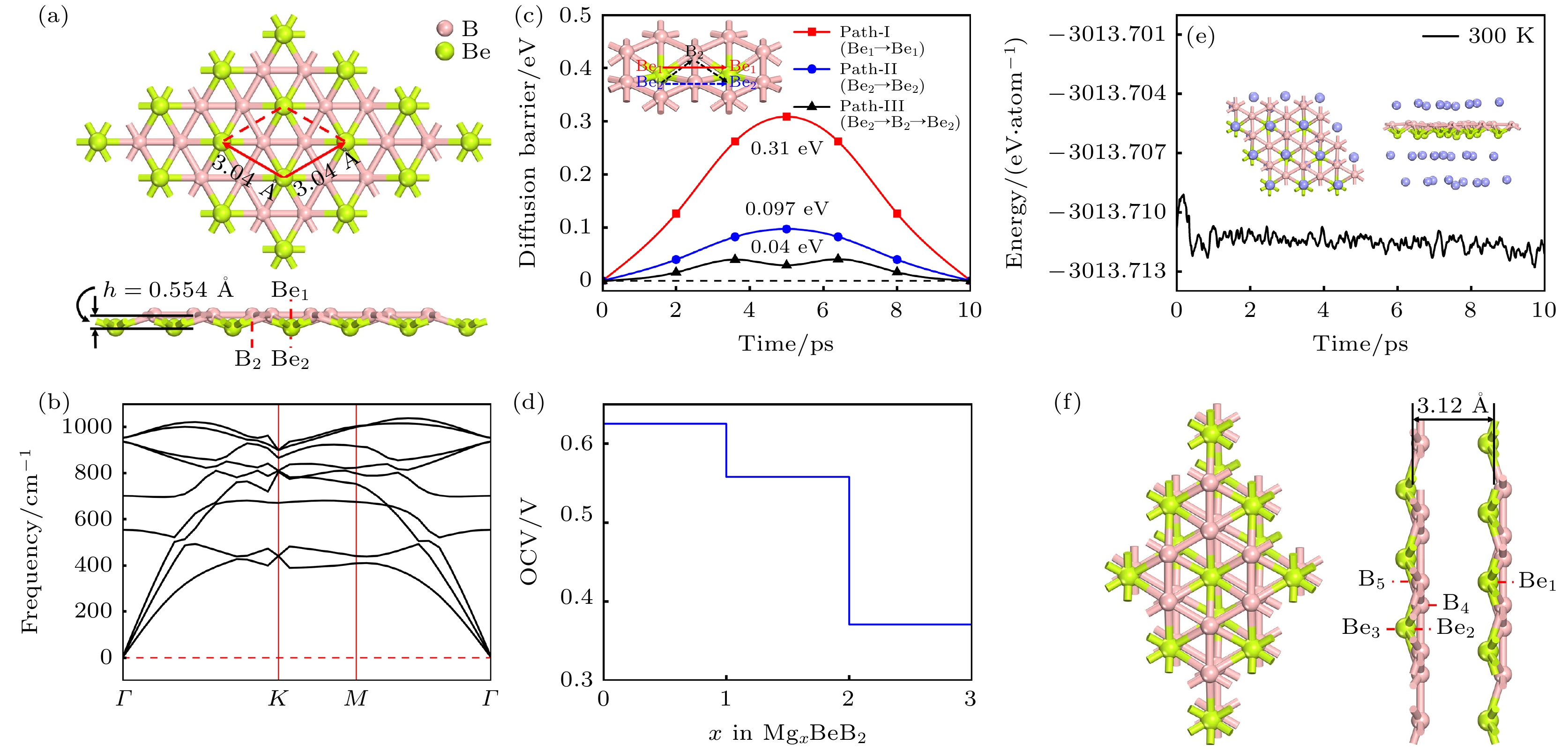
如图1(a)所示, BeB2单层结构由两个原子层组成, 其中上层B原子的排列方式呈蜂窝状, 与石墨烯结构类似, 围绕下层的Be原子环绕成六边形, 即每个Be原子与周围6个B原子配位, 每个B原子与周围3个Be原子配位. 优化完全的BeB2属于P6mm空间群, 每个初级原胞中含有两个B原子和一个Be原子, 其中原胞的晶格常数为a = b = 3.037 Å, 原子层间的高度为0.554 Å. 这与先前研究的BeB2的晶格常数(3.04 Å)和原子层间高度(0.576 Å)基本一致[10].
为了验证BeB2的稳定性, 进行了声子谱计算. 如图1(b)所示, 在布里渊区范围内, BeB2的声子谱曲线没有出现虚频, 证明该材料具有良好的动力学稳定性. 狄拉克锥的发现(图1(c))表明BeB2拥有良好的金属导电能力, 是作为金属离子电池阳极的优秀材料. 为了分析该结构导电性能的来源, 进一步分析了BeB2的分态密度图(图1(d)), B原子的p轨道以及Be的p轨道在费米能级附近贡献大部分电子.
-
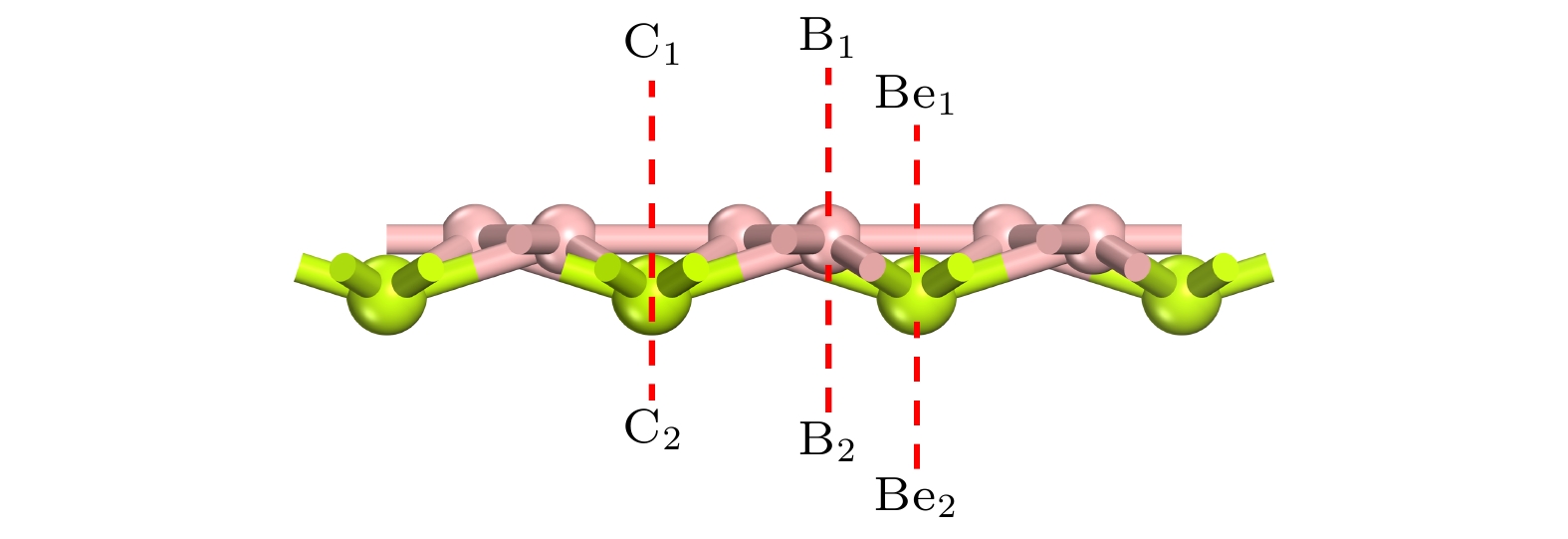
BeB2作为电池阳极材料, 需要对金属原子有一定强度的吸附作用, 从而避免充放电过程的不可逆或是产生金属团簇. 因此, 根据BeB2的结构对称性, 我们测试了6个可能的吸附点位, 如图2所示, 分别为: Be原子的上方与下方(Be1和Be2), B原子的上方与下方(B1和B2)以及B—B键的上方与下方(C1和C2). 为了研究金属原子与BeB2的相互作用, Mg原子在每个吸附点位的吸附能(
$ {E}_{{\mathrm{a}}{\mathrm{d}}{\mathrm{s}}} $ )经过下式的计算可得此式中,
$ {E}_{{\mathrm{s}}{\mathrm{u}}{\mathrm{b}}{\mathrm{s}}{\mathrm{t}}{\mathrm{r}}{\mathrm{a}}{\mathrm{t}}{\mathrm{e}}} $ 和$ {E}_{{\mathrm{M}}+{\mathrm{s}}{\mathrm{u}}{\mathrm{b}}{\mathrm{s}}{\mathrm{t}}{\mathrm{r}}{\mathrm{a}}{\mathrm{t}}{\mathrm{e}}} $ 是阳极材料吸附金属离子前后的总能量,$ {E}_{{\mathrm{M}}} $ 是金属晶体结构中每个金属原子的能量,$ n $ 表示被吸附金属离子的数量. 吸附Mg原子之后的BeB2体系经过结构完全优化后, B1和 C1点位的Mg原子转移至 Be1点位, C2点位的Mg原子转移至B2点位. 对于二维BeB2来说, 仅有三个稳定吸附点位, 其中Be1点位的吸附最稳定, 吸附能为–2.67 eV, Be2和B2点位为次稳定位, 吸附能分别为–2.24 eV和–2.16 eV.为了研究Mg原子与BeB2之间的相互作用, 计算了BeB2单层结构吸附Mg原子前后的差分电荷以及分态密度. 图3(a)和图3(b)显示了Mg2+在最稳定吸附位Be1点位的俯视和侧视差分电荷密度图, 蓝色区域代表电子积累区, 黄色区域代表电子消耗区. 由图3得知基底和Mg2+之间存在明显的电荷转移, 即Mg向BeB2转移了部分电子. Hirshfeld 电荷分析显示 Mg2+在三个稳定的吸附点位均有不同程度的电荷转移, 电荷转移量(∆Q)与吸附能的绝对值成正比, 具体数值见表1. 图3(c)和图3(d)为原始的BeB2超胞和Mg吸附在Be1位的BeB2的分态密度图, 可以发现吸附Mg2+后体系保持金属性, B原子的p轨道为体系贡献最多的电子, 同时Mg原子s轨道及Be和p轨道也贡献了部分电子, 吸附后的金属性质可有效提高电极的电导率.
-
阳极材料拥有较高的离子扩散速度能够保证电池具有较高的充放电速率, 这是作为金属离子电池的必要条件. 因此, 针对Mg2+在BeB2表面的扩散性能作了深入分析. 基于Mg2+在二维BeB2上稳定的吸附点位及BeB2的高对称性, 选择了三条可能路径, 如图4所示. 路径Ⅰ: Mg2+在最近邻的两个 Be原子上方(Be1位)扩散, 即沿 Be1-Be1方向扩散, 相应的扩散势垒为0.31 eV; 路径Ⅱ: Mg2+在最近邻的两个 Be原子下方(Be2位)扩散, 即沿 Be2-Be2方向扩散, 相应的扩散势垒为0.097 eV; 路径Ⅲ: Mg2+沿着 Be2-B2-Be2方向扩散, 其最低的扩散势垒为0.04 eV. 综上所述路径Ⅲ为最佳的扩散路径. 经过对比, Mg2+在 BeB2表面的最低扩散势垒明显低于Be2B和α-beryllene的最优路径上的扩散势垒[18].
-
为获得BeB2作为镁离子电池阳极的最大理论容量, 我们需要确定可稳定吸附Mg2+的最大数量. 因此, 采用多层吸附的方法, 使 Mg原子尽可能地占据所有稳定的吸附点位. 本文选用一个3×3×1的BeB2超级原胞, 根据 BeB2非平面的结构特点, 第一层吸附Mg2+的方式是将 Mg原子占满所有 Be1位和 Be2位. 而对于吸附第二层 Mg2+时, Mg原子仅可占据 B2位. 因此, 我们考虑了三类不同吸附浓度的构型, 相应的化学式表示为 MgBeB2, Mg2BeB2 和Mg3BeB2. 经过充分的结构弛豫, 所有构型的Mg2+均在吸附点位保持稳定吸附, 并且整个体系的结构没有出现变形, 如图5所示.
为了评估每个吸附构型上Mg2+的吸附强度, 根据(1)式计算了每个构型的平均吸附能. 如图6(a)所示, 平均吸附能随吸附Mg2+浓度的增加而升高, 主要是由于吸附Mg2+的个数增加导致Mg2+之间的相互排斥作用, 进而削弱了体系的吸附能. 为了研究每个吸附构型的稳定性, 进行了形成能的计算:
$ {E}_{{{\mathrm{M}}{\mathrm{g}}}_{x}{\mathrm{B}}{\mathrm{e}}{{\mathrm{B}}}_{2}} $ 代表 BeB2 吸附Mg之后的总能量, x表示吸附的Mg离子的浓度 (1—3),$ {E}_{{\mathrm{B}}{\mathrm{e}}{{\mathrm{B}}}_{2}} $ 表示纯的BeB2 结构的总能量,$ {E}_{{\mathrm{M}}{\mathrm{g}}} $ 是体相中每个Mg原子的能量. 计算结果如图6(b)所示, 三个吸附体系得出的形成能均落在凸包图曲线上, 表明了三个吸附浓度的结构都是热力学稳定的. 根据计算的电子局域函数(ELF)图(图6(c))显示, 吸附一层和两层Mg2+的周围均呈现局域化的电子, 形成了带负电的电子云, 有利于Mg2+的吸附.采用以下公式计算理论容量:
式中, z 为吸附的金属离子价电子数; x 为吸附的Mg2+数量; F 是法拉第常数(26801 mA·h·mol–1), M 为阳极材料原胞的摩尔质量. 由(3)式得出 BeB2的最大理论容量为 5250 mA·h·g–1, 和α-beryllene (5956 mA·h·g–1)有着相当的理论容量[18]. 为了研究BeB2作为镁离子电池阳极时由于充放电所引起的体积变化, 进一步计算了Mg3BeB2相比BeB2的体积变化量, 得到的最大体积膨胀率约为2%, 表明二维阳极 BeB2具有良好的可逆性.
开路电压是评估金属离子电池阳极材料性能的一个重要指标, 其可以根据下式来计算:
式中,
$ {E}_{{{\mathrm{M}}}_{{x}_{1}}} $ 和$ {E}_{{{\mathrm{M}}}_{{x}_{2}}} $ 分别表示阳极吸附离子浓度为$ {x}_{1} $ 和$ {x}_{2} $ 时的总能;$ {E}_{{\mathrm{M}}} $ 是金属体相结构中的每个金属原子的能量; z表示所吸附金属原子的价电子数; e为电子电量. 经过计算得到的BeB2在各个吸附的Mg2+浓度下的开路电压如图6(d)所示, 电压窗口的范围为在0.37—0.62 V, 平均开路电压为 0.33 V, 与Be2B(0.29 V)[9]及α-beryllene (0.24 V)[18]作为镁离子电池阳极的平均电压相当, 可以结合高电压阴极提供较高的功率密度. 基于最大浓度的吸附体系(Mg3BeB2), 进一步评估了其动力学稳定性. 经过分子动力学模拟分析, 结果如图6(e), 在温度为 300 K时, Mg3BeB2体系经过10 ps的动力学模拟之后结构保持平稳, BeB2平面出现小幅度的变化, 所有 Mg2+在平衡位置仅有较小的偏移, 总能量在较小的范围内波动, 表明在室温条件下 BeB2储存 Mg2+之后能够保持结构稳定. -
上文中, 我们只考虑镁在单层BeB2上的吸附和扩散. 在实际应用中, 仅仅使用单层材料作为金属离子的电极具有很大的难度. 因此, 研究了Mg在双层BeB2中的吸附和扩散性能. 双层BeB2由两个3×3×1的单层BeB2超胞堆叠而成, 考虑其结构特点, 尝试了图7中的三种堆叠方式, 分别命名为AA堆叠(上层Be原子与下层Be原子重叠), AB堆叠(上层Be原子与下层B原子重叠)和AC堆叠(上层Be原子和下层B—B键重叠).
为了验证双层BeB2的最稳定结构, 利用(5)式计算了双层BeB2的层间结合能(
${E_{\text{b}}}$ ):式中,
${E_{{\text{Bilayer-Be}}{{\text{B}}_{2}}}}$ 是双层BeB2的总能量;${E_{{\text{Be}}{{\text{B}}_{2}}}}$ 是单层BeB2的总能量; n是双层BeB2中的总原子数. 如图8(a)所示, 通过计算层间结合能随双分子层间距的变化, 确定了三种堆叠方式的最稳定结构. AB堆叠的层间结合能为–120.97 meV/atom, 小于AA堆叠(–83.21 meV/atom)和AC堆叠(–104.15 meV/atom), 是最稳定的堆叠方式, 其层间距为3.12 Å.根据双层BeB2的对称性, 我们考虑了三种吸附区域: 1) Mg吸附在双层BeB2的上层; 2) Mg嵌入双层BeB2的中间区域; 3) Mg吸附在双层BeB2的下层. 通过充分结构优化如图8(b)所示的一系列吸附点位, 得到Mg吸附在上层的Be1(Eads = –2.24 eV)、中层的B4(Eads = –1.90 eV)和下层的B5(Eads = –1.38 eV)时最稳定. 与单层BeB2相比, 双层BeB2在稳定点位的吸附能有所增大, 即意味着对金属原子的吸附强度有所减弱.
根据双层BeB2的稳定吸附点位, 设计了图9中的5条扩散路径并计算了相应的扩散势垒. 在双层BeB2的上层, 由于只存在一个最稳定点位(Be1), 只考虑Be1-Be1(路径Ⅰ)一条路径, 计算出扩散势垒为0.84 eV. 而在双层BeB2的中层和下层, 除了存在最稳定点位(B4, B5)外, 还分别存在一个次稳定点位(Be2, Be3), 因此计算了4条扩散路径, 即B4-B4(路径Ⅱ), B4-Be2-B4(路径Ⅲ), B5-B5(路径Ⅳ)和B5-Be3-B5(路径Ⅴ), 其扩散势垒分为别0.43, 0.33, 0.135和0.134 eV. 由图9可以看出, Mg更容易在双层BeB2的下层发生扩散, 但是扩散势垒明显高于Mg在单层的BeB2的扩散势垒.
-
本文系统地研究了BeB2单层结构作为镁离子电池阳极的潜力, 并将其与几种镁离子阳极材料进行对比, 如表2所列. 综合分析几种材料的性能, BeB2可以稳定吸附Mg2+, 且其扩散势垒(0.04—0.31 eV)低于表中所列举的其他材料, 这预示BeB2具有更快的充放电速率; BeB2的理论容量(5250 mA·h·g–1)高于表中大部分材料, 接近二维材料α-beryllene(5696 mA·h·g–1), 但低于Be2B (7436 mA·h·g–1); BeB2的平均开路电压(0.33 V)与二维材料α-beryllene (0.24 V)和Be2B (0.29 V)相当, 但低于表中的其他材料, 可以与高电压阴极相结合, 提供较高的功率密度; 另外, BeB2体积膨胀率(2%)仅高于二维材料α-beryllene (–0.18%), 低体积膨胀代表在电池中拥有较好的可逆性和更长的使用寿命. 除了理论研究的性能对比之外, 我们还对比分析了实验上合成的一些材料的性能, 如表2所列. 综合理论容量以及开路电压的对比, 可以验证本文中理论计算的可信度. 因此, 基于以上优异的性能, 我们认为BeB2具有作为性能均衡的镁离子电池阳极材料潜在应用价值.
-
本文利用第一性原理对BeB2作为镁离子电池阳极展开了系统性研究, Mg2+可以稳定地吸附于BeB2的表面, 并且吸附Mg2+的BeB2展现出良好的导电性. 通过计算发现BeB2的理论容量为5250 mA·h·g–1, 这是影响电池性能的重要因素之一. Mg2+在BeB2表面的最低扩散势垒为0.04 eV, 极低的扩散势垒意味着可以为电池带来更高的充放电速率. 此外BeB2展示了合适的平均开路电压(0.33 V), 较低的体积膨胀率(2%). 基于实际应用的考量, 我们构建了双层BeB2, 研究了Mg在双层结构中的吸附和扩散行为. 基于以上研究结果, 我们认为BeB2是一种性能均衡且非常优异的镁离子电池阳极材料, 具有广阔的发展前景.
二维BeB2作为镁离子电池阳极材料的理论研究
Theoretical study of two-dimensional BeB2 monolayer as anode material for magnesium ion batteries
-
摘要: 为了加快镁离子电池的开发与应用, 寻找合适的镁离子电池阳极材料势在必行. 此外, 具有较低摩尔质量的阳极材料有利于获得较高的理论存储容量. 因此, 本文采用基于密度泛函理论的第一性原理计算系统地研究了BeB2单层材料作为镁离子电池阳极的潜力. 计算结果表明, 基于声子谱检测, BeB2结构展现了优异的动力学稳定性. 此外, 从BeB2的能带结构可以看到清晰的狄拉克锥, 表明其具有良好的导电性能. BeB2可以稳定吸附镁离子, 并且镁离子在该材料上表现了较低的扩散势垒 (0.04 eV), 这意味着更快的充放电速率. 重要的是, BeB2展现了超高的理论容量 (5250 mA·h·g–1)、较低平均开路电压 (0.33 V)以及较小的体积膨胀 (2%). 此外, Mg离子在双层BeB2结构中的吸附能为–1.38 — –2.24 eV, 扩散势垒为0.134 — 0.84 eV. 综合以上性能, 我们相信BeB2可以作为一种优秀的镁离子电池阳极材料.Abstract:
Rechargeable lithium-ion batteries as the main energy storage equipment should possess high power density, excellent reversible capacity, and long cycle life. However, due to the high cost and dendrite growth of Li, searching for non-Li-ion batteries is urgent. Compared with lithium, magnesium has abundant resources, small ionic radius, and high energy density. Therefore, magnesium-ion batteries (MIBs) can serve as the next generation metal-ion batteries. Two-dimensional materials based on Be or B element acting as the anode of metal-ion batteries always exhibit high theoretical storage capacity. Using first-principles calculations, we systematically explore the potential of BeB2 as MIBs anode. The optimized BeB2 monolayer structure shown in Fig. (a) consists of two atomic layers, where each Be atom is coordinated with six B atoms, and each B atom is coordinated with three Be atoms. The lattice constants are a = b = 3.037 Å with a thickness of 0.554 Å. From the phonon spectrum calculations, the absence of imaginary modes indicates the dynamic stability of BeB2 monolayer. The presence of a Dirac cone further suggests the excellent conductivity (Fig.(b)). Three stable adsorption sites (Be1: top of Be atoms; Be2 and B2: bottom of Be and B atoms) are labeled in Fig. (a). Taking symmetry into account, we consider three pathways to evaluate the migration of Mg atom on BeB2 monolayer (Fig.(c)). The corresponding lowest diffusion energy barrier is 0.04 eV along Path III. The stable configuration with the maximum adsorption Mg concentration is shown in Fig.(d), which generates a theoretical capacity of 5250 mA·h·g–1. The calculated average open-circuit voltage is 0.33 V. Based on ab initio molecular dynamics simulations, the total energy of BeB2, with Mg adsorbed, fluctuates within a narrow range, suggesting that BeB2 can sustain structural stability after storing Mg at room temperature (Fig.(e)). Finally, for practical application, we investigate the adsorption and diffusion behavior of Mg on bilayer BeB2. Three configurations are considered: AA stacking (overlapping of Be atoms in upper layer with Be atoms in lower layer), AB stacking (overlapping of Be atoms in upper layer with B atoms in lower layer), and AC stacking (overlapping of Be atoms in upper layer with B—B bonds in lower layer). The most stable configuration is AB stacking (shown in Fig.(f)) with the interlayer spacing of 3.12 Å and the binding energy of –120.97 meV/atom. Comparing with the BeB2 monolayer structure, the adsorption energy of Mg is –2.24 eV for Be1, –1.38 eV for B5 site, and –1.90 eV for B4 site, while the lowest diffusion energy barrier is 0.13 eV along the path of B5-Be3-B5. Therefore, according to the above-mentioned properties, we believe that BeB2 monolayer can serve as an excellent MIBs anode material. -
Key words:
- magnesium-ion battery /
- two-dimensional materials /
- first-principles calculations .
-

-
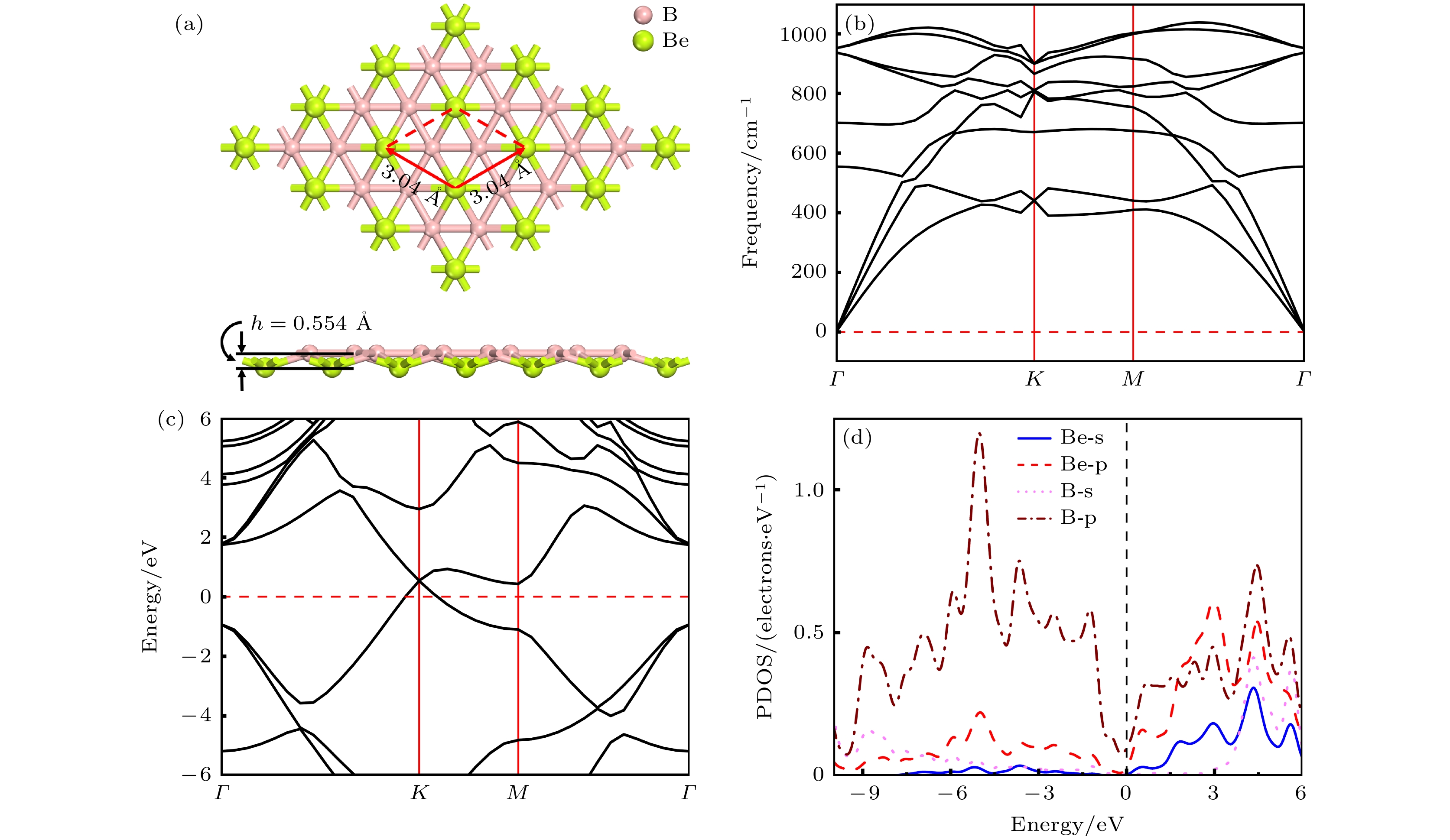
图 1 (a) BeB2单层结构的俯视图及侧视图, 红色框架展示了BeB2单层的初级原胞结构, h表示 BeB2单层的厚度; BeB2单层的声子谱(b)、能带结构(c)以及分态密度图(d)
Figure 1. (a) Top and side views of BeB2 monolayer. Red frame represents the primary cell structure of BeB2, and h represents the thickness of BeB2 monolayer. (b) Phonon spectra, (c) electronic band structures, (d) partial density of states of BeB2 monolayer.
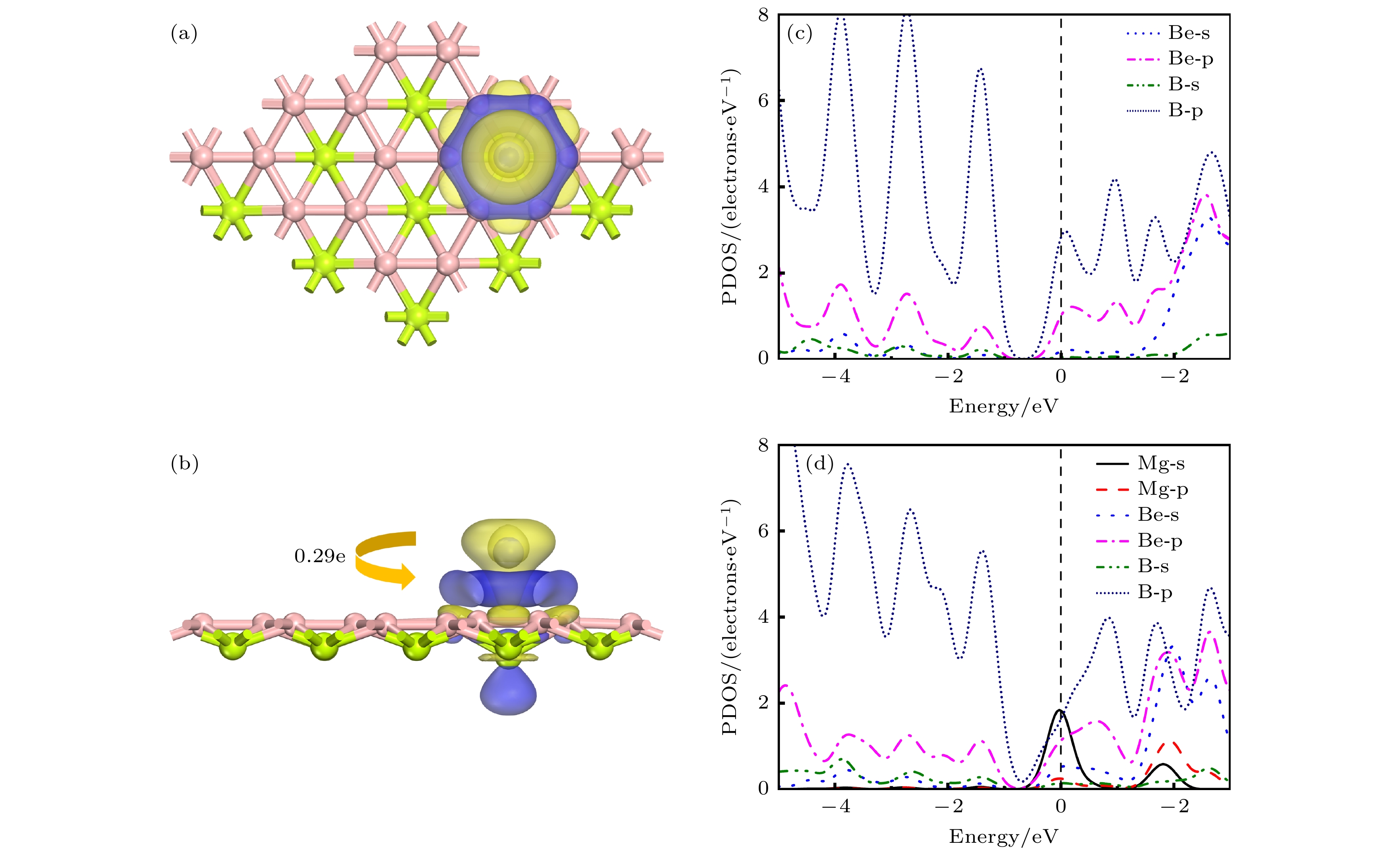
图 3 Mg2+吸附在 Be1位的差分电荷密度分布俯视图 (a)和侧视图 (b); BeB2单层(c)以及Mg2+吸附在Be1位的BeB2 (d)的分态密度图
Figure 3. Top (a) and side (b) views of charge density difference for Mg2+ absorbed at Be1 site; partial density of states of BeB2 monolayer (c) and BeB2 with Mg2+ adsorbed at Be1 site (d).
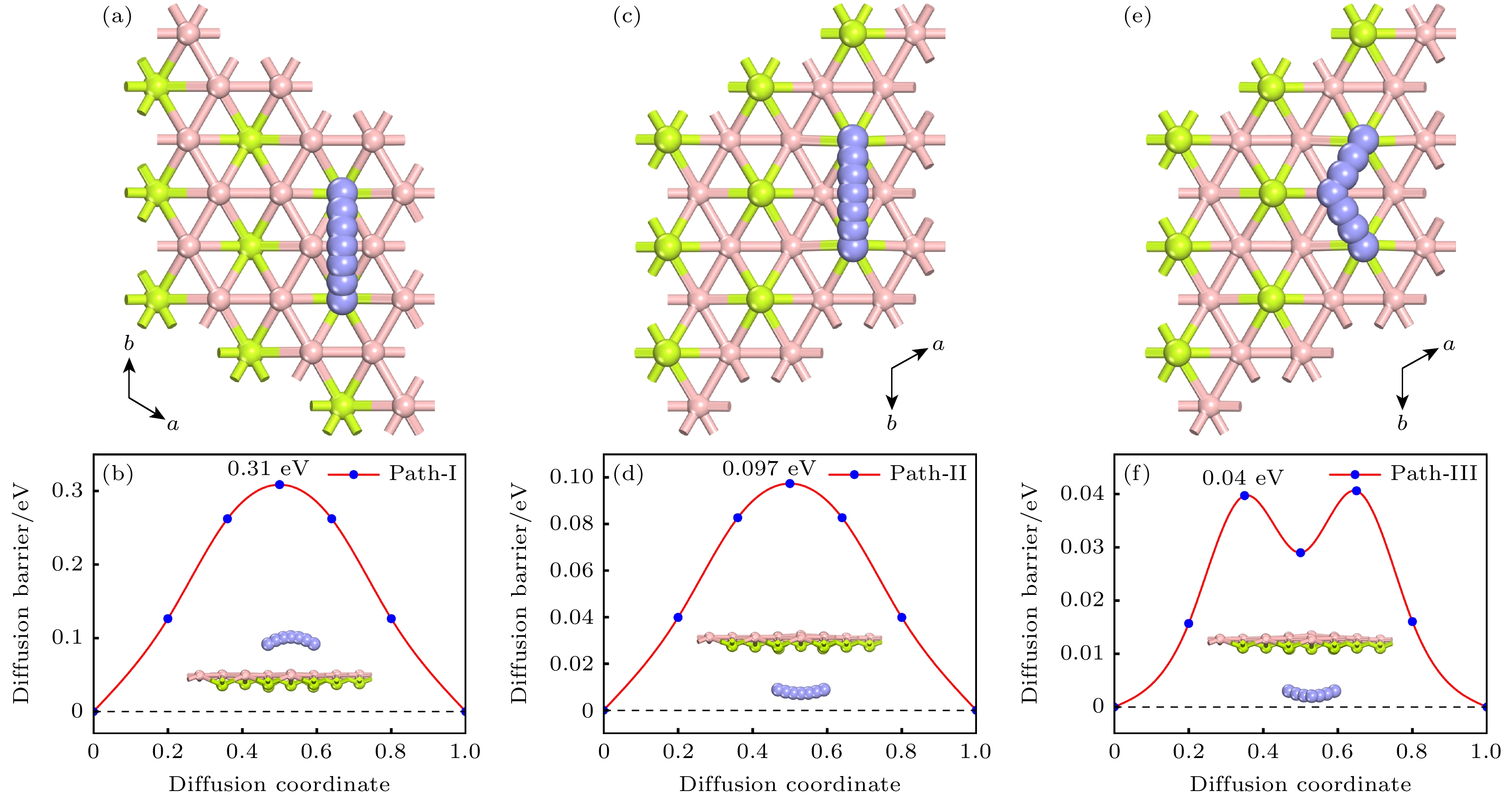
图 4 Mg2+在 BeB2表面的扩散路径俯视图, 分别对应路径 Ⅰ (a), II (c), Ⅲ (e); 对应路径的扩散势垒 路径Ⅰ (b), II (d), Ⅲ (f). 插图为 Mg2+扩散的侧视图
Figure 4. (a), (c), (e) Top views of Mg2+ diffusion paths on BeB2 surface, corresponding to path Ⅰ, II, and Ⅲ, respectively; (b), (d), (e) diffusion barriers of path Ⅰ, II, and Ⅲ. Insets are side views of Mg2+ diffusion pathways.
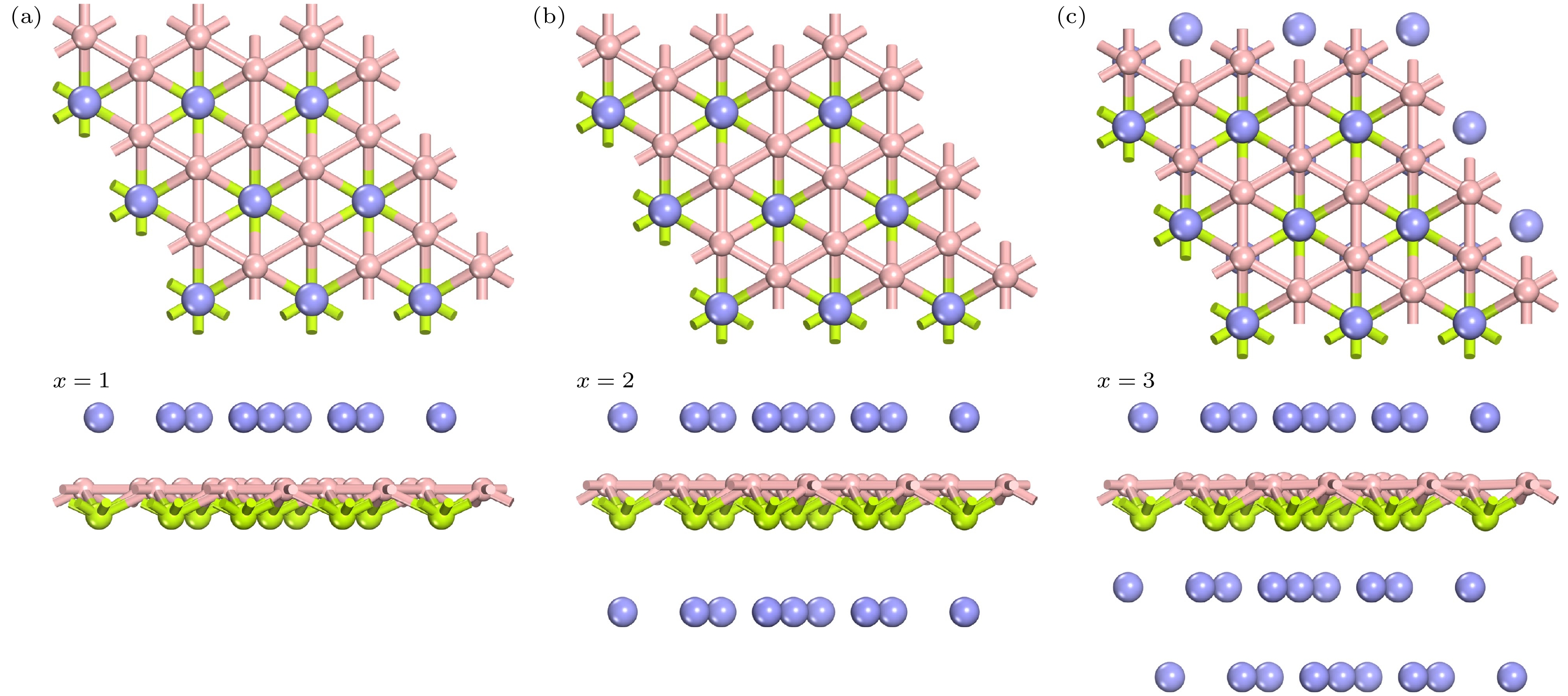
图 5 不同吸附浓度的MgxBeB2体系完全优化后的俯视图和侧视图 (a) MgBeB2; (b) Mg2BeB2; (c) Mg3BeB2
Figure 5. Top and side views of different configurations after full optimization: (a) MgBeB2; (b) Mg2BeB2; (c) Mg3BeB2.
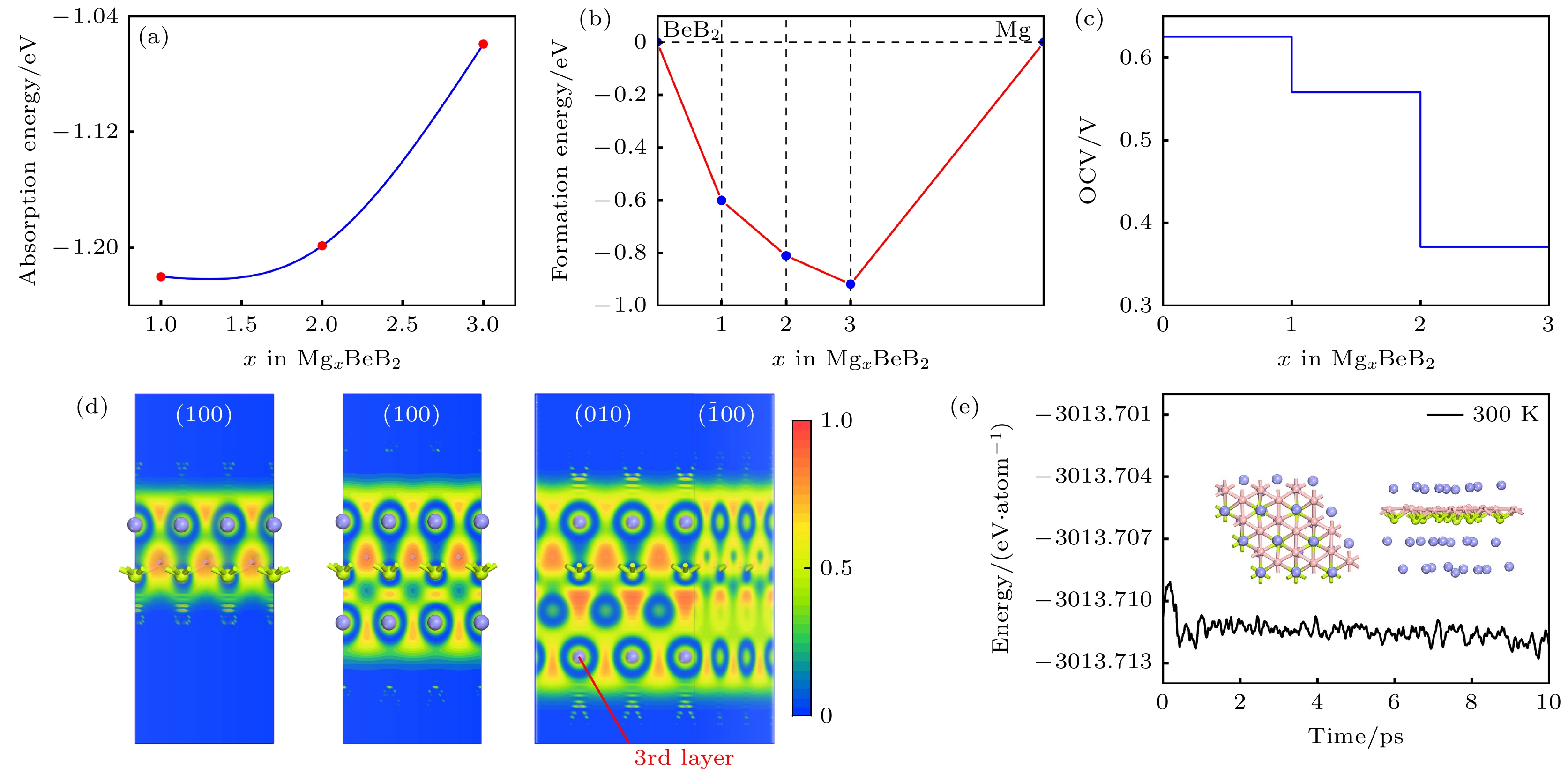
图 6 (a) 平均吸附能随吸附浓度变化的曲线; (b) MgxBeB2形成能曲线; (c) MgBeB2, Mg2BeB2和 Mg3BeB2的ELF侧视图; (d) 吸附Mg2+的BeB2单层的开路电压; (e) Mg3BeB2在 300 K条件下分子动力学模拟 10 ps 后的能量曲线, 插图为模拟结束时体系的俯视图及侧视图
Figure 6. (a) Variation of average adsorption energy with the concentration of adsorbed Mg; (b) formation energy of MgxBeB2; (c) side views of electron function localization for MgBeB2, Mg2BeB2 and Mg3BeB2, respectively; (d) OCV of BeB2 monolayer with different Mg concentration; (e) total energy variation of Mg3BeB2 during ab initio molecular dynamics at 300 K. Inset exhibits the top and side snapshots at the end of 10 ps.
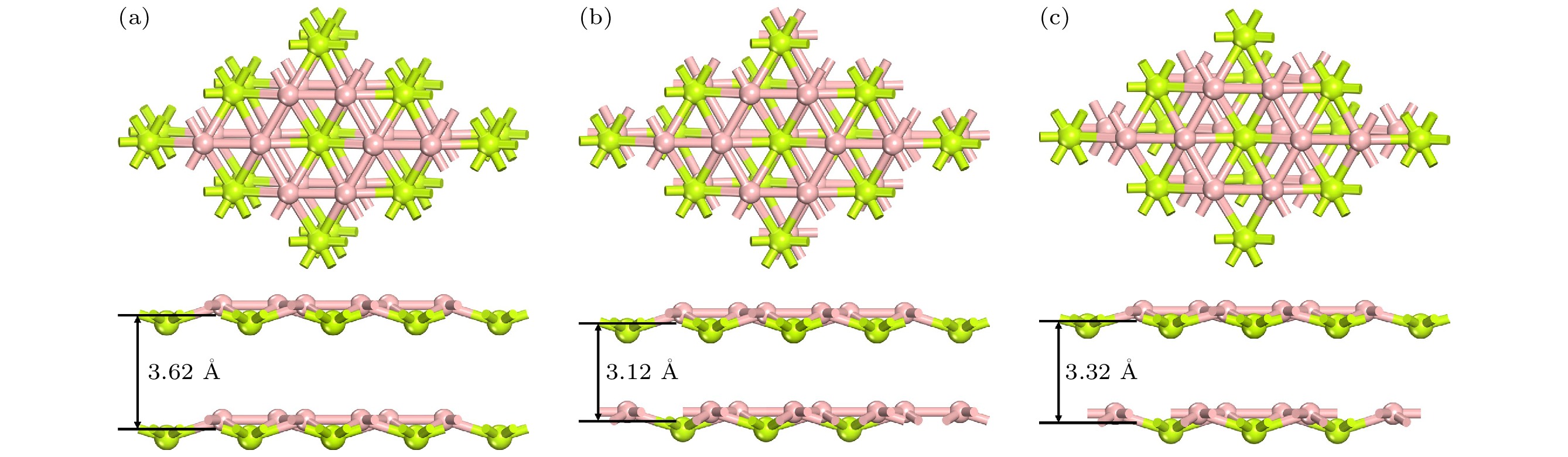
图 7 (a) AA, (b) AB和(c) AC堆叠的双层BeB2的俯视图和侧视图
Figure 7. Top and side views of bilayer BeB2 with stacking of (a) AA, (b) AB, and (c) AC.
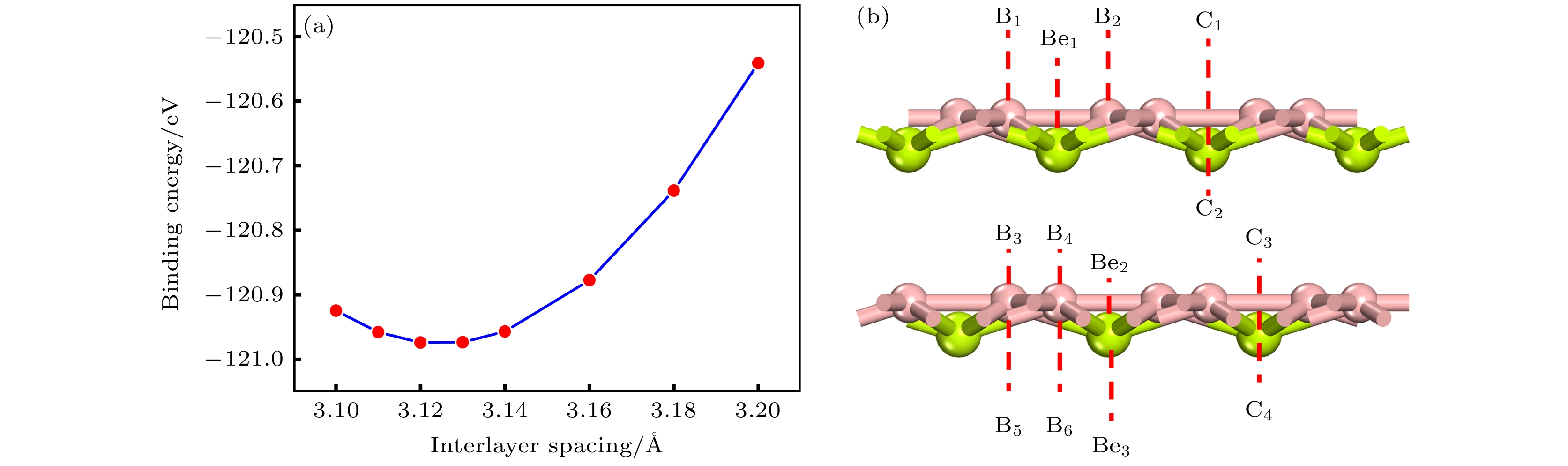
图 8 (a) AB堆叠的双层BeB2层间结合能随双分子层间距的变化曲线; (b) AB堆叠的双层BeB2所有可能的吸附点位
Figure 8. (a) The variation of interlayer binding energy of bilayer BeB2 in AB stacking mode with interlayer spacing; (b) all possible adsorption sites of bilayer BeB2 in AB stacking mode.
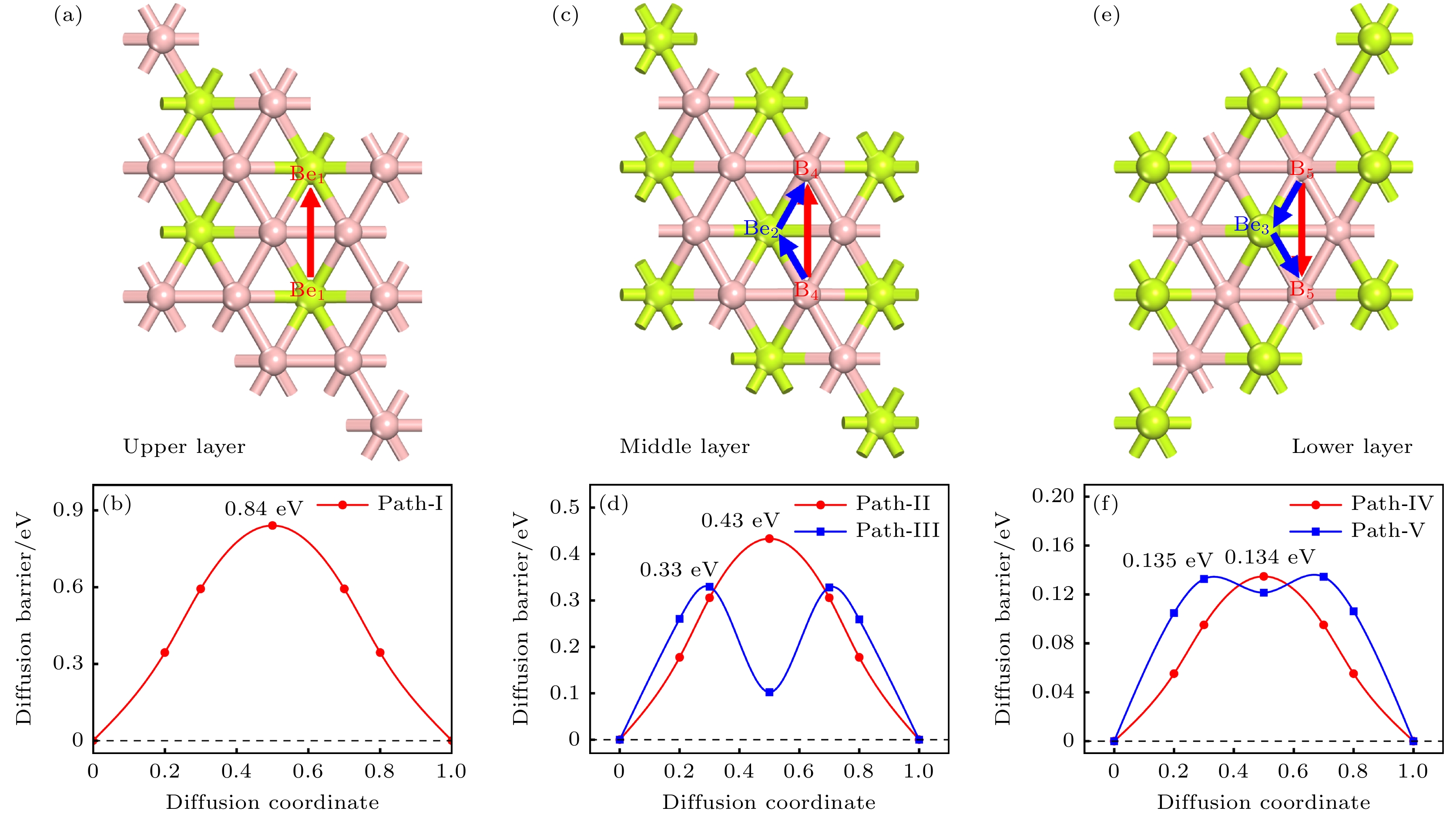
图 9 双层BeB2各层的扩散路径俯视图, 分别对应上层 (a)、中层(c)、下层(e), 以及对应路径的扩散势垒: 上层(b)、中层(d)、下层(f)
Figure 9. Top view of diffusion paths in each region of bilayer BeB2 in AB stacking, corresponding to the upper layer (a), middle layer (c), and lower layer (e); the diffusion barriers corresponding to the paths: upper layer (b), middle layer (d), and lower layer (f).
表 1 Mg2+在 BeB2表面上不同吸附点位的吸附能及转移电荷数量
Table 1. Adsorption energy at different sites and the charge transfers of Mg2+ on BeB2 monolayer.
Adsorption site Eads/eV ΔQ/e Be1 –2.67 0.29 Be2 –2.24 0.20 B2 –2.16 0.18  下载: 导出CSV
下载: 导出CSV
表 2 几种镁离子电池阳极材料与BeB2的性能对比 (*标识为实验数据)
Table 2. Comparison of several two-dimensional materials with BeB2 as MIBs anode (*experimental results).
阳极材料 吸附能/eV 扩散势垒/eV 理论容量/(mA·h·g–1) 平均开路电压/V 体积膨胀率/% BeB2 –2.67 0.04 5250 0.33 2 β12 borophene[6] –0.696 0.97 2480 — — χ3 borophene[6] –0.199 — 2400 — — Be2B[9] –0.7 0.1 7436 0.29 0.3 α-beryllene[18] –0.24 0.099 5956 0.24 –0.18 Si2BN[19] –1.22 0.08 648 0.67 — BSi[20] –2.34 0.86 2749 0.84 — Arsenene[21] 2.48 0.21 1429 0.83 <16 B-MoS2[22] –0.024 0.6 921 0.154 2.67 B40[23] — 0.20 744 5.5 — *TiP2O7[24] — 0.62 72* 2.4* 3.2* *Ge57Bi43[25] — — 847.5* 0.32—0.35* — Ge[26] — 0.7 1476 0.241 –178 *Mg2Ga5[27] — — 290* 0.01—0.7* — *Nano-Bi[28] — — 350* 0—0.25* — *石墨[29] — — 22* 0.15* — *RGO/Bi[30] — — 372* 0.25* —
下载: 导出CSV
-
[1] Perveen T, Siddiq M, Shahzad N, Ihsan R, Ahmad A, Shahzad M I 2020 Renewable Sustainable Energy Rev. 119 109549 doi: 10.1016/j.rser.2019.109549 [2] Ma Y, Doeff M M, Visco S J, Jonghe L C D 1993 J. Electrochem. Soc. 140 2726 doi: 10.1149/1.2220900 [3] Hwang J Y, Myung S T, Sun Y K 2017 Chem. Soc. Rev. 46 3529 doi: 10.1039/C6CS00776G [4] Rajagopalan R, Tang Y G, Ji X B, Jia C K, Wang H Y 2020 Adv. Funct. Mater. 30 1909486 doi: 10.1002/adfm.201909486 [5] Lin J Y, Yu T, Han F J J, Yang G C 2020 Wiley Interdiscip. Rev. Comput. Mol. Sci. 10 e1473 doi: 10.1002/wcms.1473 [6] Mortazavi B, Rahaman O, Ahzi S, Rabczuk T 2017 Appl. Mater. Today 8 60 doi: 10.1016/j.apmt.2017.04.010 [7] Yeoh K H, Chew K H, Chu Y Z, Yoon T L, Rusi, Ong D S 2019 J. Appl. Phys. 126 125302 doi: 10.1063/1.5110225 [8] Ullah S, Denis P A, Sato F 2017 Appl. Mater. Today 9 333 doi: 10.1016/j.apmt.2017.08.013 [9] Ye X J, Gao Q, Cao H B, Wang X H, Liu C S 2023 Appl. Phys. Lett. 122 223902 doi: 10.1063/5.0153381 [10] Wan M Q, Zhao S Q, Zhang Z Y, Zhou N G 2022 J. Phys. Chem. C 126 9642 doi: 10.1021/acs.jpcc.2c02563 [11] Wu Y, Hou J 2022 Phys. Chem. Chem. Phys. 24 14953 doi: 10.1039/D2CP00690A [12] Segall M D, Lindan P J D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C 2002 J. Phys. Condens. Matter 14 2717 doi: 10.1088/0953-8984/14/11/301 [13] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 doi: 10.1103/PhysRevLett.77.3865 [14] Zhang G X, Tkatchenko A, Paier J, Appel H, Scheffler M 2011 Phys. Rev. Lett. 107 245501 doi: 10.1103/PhysRevLett.107.245501 [15] Tkatchenko A, Scheffler M 2009 Phys. Rev. Lett. 102 073005 doi: 10.1103/PhysRevLett.102.073005 [16] Govind N, Petersen M, Fitzgerald G, King-Simith D 2003 Comput. Mater. Sci. 28 250 doi: 10.1016/S0927-0256(03)00111-3 [17] Henkelman G, Jónsson H 2000 J. Chem. Phys. 113 9978 doi: 10.1063/1.1323224 [18] Gao Q, Ye X J, Liu C S 2023 Phys. Chem. Chem. Phys. 25 6519 doi: 10.1039/D2CP04433A [19] Panigrahi P, Mishra S B, Hussain T, Nanda B. R. K, Ahuja Rajeev 2020 ACS Appl. Nano Mater. 3 9055 doi: 10.1021/acsanm.0c01747 [20] Xiao C, Tang X Q, Peng J F, Ding Y H 2021 Appl. Surf. Sci. 563 150278 doi: 10.1016/j.apsusc.2021.150278 [21] Khan A A, Muhammad I, Ahmad R, Ahmad L 2021 Ionics 27 4819 doi: 10.1007/s11581-021-04239-y [22] Wu D H, Yang B C, Zhang S R, Ruckenstein E Chen H Y 2021 J. Colloid Interface Sci. 597 401 doi: 10.1016/j.jcis.2021.04.002 [23] Shakerzadeh E, Kazemimoghadam F 2021 Appl. Surf. Sci. 538 148060 doi: 10.1016/j.apsusc.2020.148060 [24] Xiong F Y, Jiang Y L, Cheng L, Yu R H, Tan S S, Tang C, Zuo C L, An Q Y, Zhao Y L, Gaumet J J, Mai L Q 2022 Interdiscip. Mater. 1 140 doi: 10.1002/idm2.12004 [25] Zhang Z H, Song M J, Si C H, Cui W R, Wang Y 2023 eScience 3 100070 doi: 10.1016/j.esci.2022.07.004 [26] Malyi O. I, Tan T. L, Manzhos S 2013 J. Power Sources 233 341 doi: 10.1016/j.jpowsour.2013.01.114 [27] Wang L, Welborn S S, Kumar H, Li M N, Wang Z Y, Shenoy V B, Detsi E 2019 Adv. Energy Mater. 9 1902086 doi: 10.1002/aenm.201902086 [28] Shao Y Y, Gu M, Li X Y, Nie Z M, Zuo P J, Li G S, Liu T B, Xiao J Cheng Y W, Wang C M, Zhang J G, Liu J 2014 Nano Lett. 14 255 doi: 10.1021/nl403874y [29] God C, Bitschnau B, Kapper K, Lenardt C, Schmuck M , Mautner F, Koller S 2017 RSC Adv. 7 14168 [30] Penki T R, Valurouthu G, Shivakumara S, Sethuraman V A, Munichandraiah N 2018 New J. Chem. 42 5996 doi: 10.1039/C7NJ04930G -
计量
- 文章访问数: 489
- HTML全文浏览数: 489
- PDF下载数: 8
- 施引文献: 0